Alpha-1 antitrypsin deficiency
In the lungs a protein called elastin helps lung tissue expand and
contract while maintaining the shape of the airway. In response to
stressors such as infection or chemical irritants, e.g. cigarette smoke,
neutrophils produce elastase a broadly functioning proteolytic enzyme
which can help destroy bacteria. While this is beneficial to destroying
the bacteria, it also destroys the elastin in the lungs which can lead
to emphysema. To avoid this the liver produces a protein which inhibits
the function elastase called Alpha-1 antitrypsin (AAT) which is
transported to the lungs via the blood stream. If AAT is deficient,
damage occurs to the elastic fibers in the lungs resulting in emphysema
(damage to the alveoli). This can be due to external factors such as
free radicals in cigarette smoke which inhibit AAT function, or due to
genetic deficiencies.
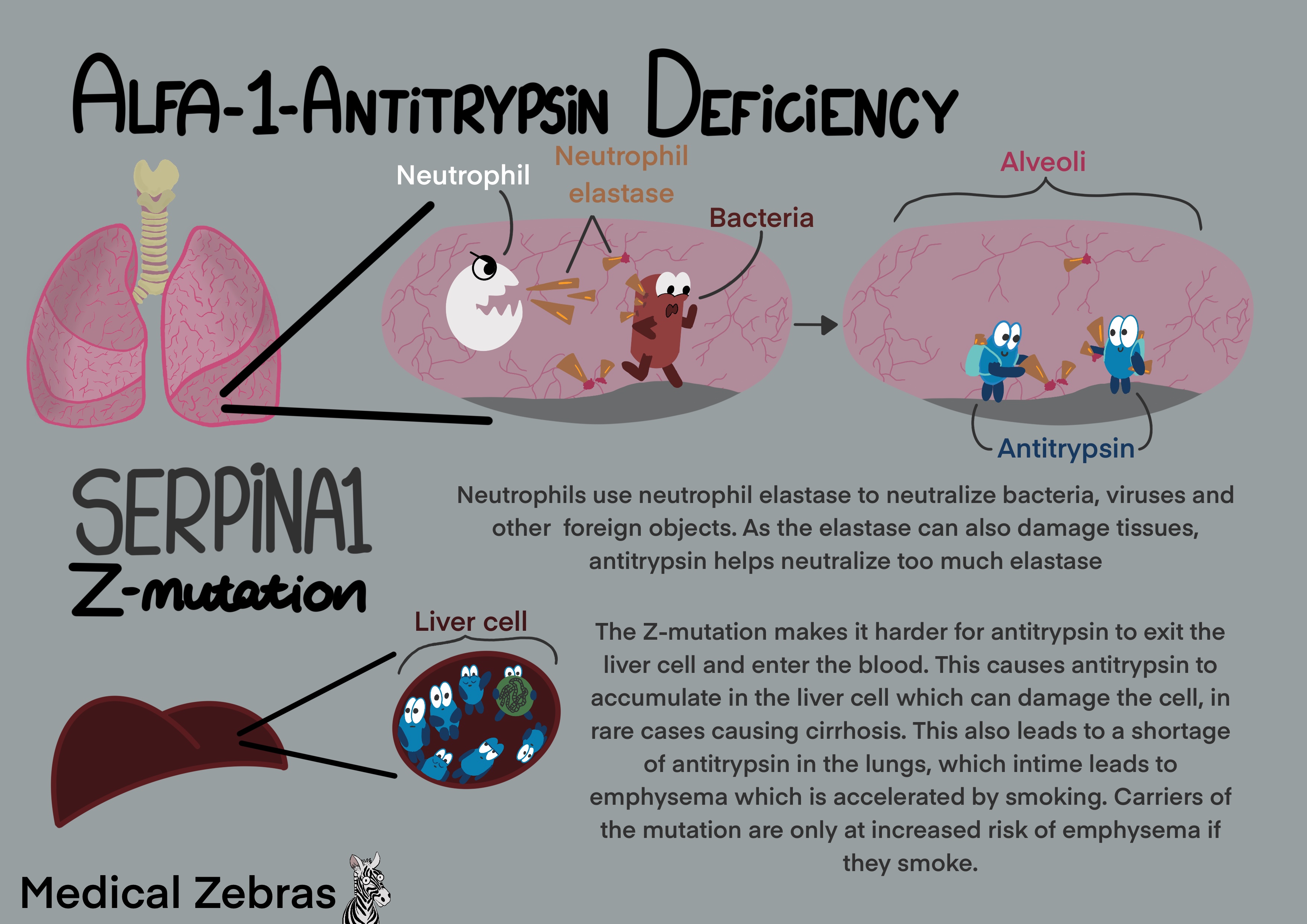
Alpha-1 antitrypsin deficiency is a autosomal recessive disorder caused by a mutation in a gene called SERPINA1. SERPINA1 is located on the long arm (q) of chromasome 14 and codes for AAT. There are many known pathogenic mutations in the SERPINA1 gene, and they have been given names of the alphabet depending on how fast their protein product travels through a gel during electrophoresis compared to the others. Two of them are especially common in the Western World, namly "Z" and "S". In the literature they are donoted as Protease inhibitor (Pi) followed by letter indicating the present mutation.
The "Z" mutation causes AAT entrapment in the liver cells leading to insufficient levels of AAT the blood stream. This accumulation can, with time, cause liver damage and in rare cases cirrhosis can occur. Some Pi*ZZ Children can develop significant liver damage requiring liver transplant(~2%). This entrapment can be observed as elevated liver enzymes in the blood tests of individuals homozygous for the Z mutation. Pi*ZZ individuals will develope emphysema via the mechanism outlined above, most over 40 years of age, but sooner if they smoke. Those who are heterozygotes for a pathogenic mutation in SERPINA1 (carriers), produce less AAT. The amount they produce is above the threshold required to protect their lungs, however, they are at increased risk for developing emphysema if they smoke.
Homozygotes for the mutation "S", were once assumed to have the same risks as homozgotes for "Z", but recent research has shown that the production of AAT in most cases does not fall below the protective threshold and will therefore not cause symptoms unless their risk is increased by smoking, their risk is therefore nearly the same as if they were carriers of the Z allele. The "S" mutation does not cause the AAT to clog up the liver cells and therefore does not cause liver cell damage like the "Z" mutation does. About 10% of persons that are compound heterozygote for the "Z" and "S" mutations(Pi*ZS) have AAT levels below the protective threshold and are therefore at risk at developing emphysema. All P*ZS are considered to be at increased risk if they smoke.
One mutation in the SERPINA1 gene, is called the "F" mutation. This mutation does not affect the amount of AAT that the liver produces, but is defective. Meaning that these individuals will have normal amounts of serum AAT.
Both infections and high amounts of estrogen (for example pregnancy) can raise the level of serum AAT. It is therefore important to take these two things into consideration when measuring serum AAT.
Alpha-1 antitrypsin deficiency is a autosomal recessive disorder caused by a mutation in a gene called SERPINA1. SERPINA1 is located on the long arm (q) of chromasome 14 and codes for AAT. There are many known pathogenic mutations in the SERPINA1 gene, and they have been given names of the alphabet depending on how fast their protein product travels through a gel during electrophoresis compared to the others. Two of them are especially common in the Western World, namly "Z" and "S". In the literature they are donoted as Protease inhibitor (Pi) followed by letter indicating the present mutation.
The "Z" mutation causes AAT entrapment in the liver cells leading to insufficient levels of AAT the blood stream. This accumulation can, with time, cause liver damage and in rare cases cirrhosis can occur. Some Pi*ZZ Children can develop significant liver damage requiring liver transplant(~2%). This entrapment can be observed as elevated liver enzymes in the blood tests of individuals homozygous for the Z mutation. Pi*ZZ individuals will develope emphysema via the mechanism outlined above, most over 40 years of age, but sooner if they smoke. Those who are heterozygotes for a pathogenic mutation in SERPINA1 (carriers), produce less AAT. The amount they produce is above the threshold required to protect their lungs, however, they are at increased risk for developing emphysema if they smoke.
Homozygotes for the mutation "S", were once assumed to have the same risks as homozgotes for "Z", but recent research has shown that the production of AAT in most cases does not fall below the protective threshold and will therefore not cause symptoms unless their risk is increased by smoking, their risk is therefore nearly the same as if they were carriers of the Z allele. The "S" mutation does not cause the AAT to clog up the liver cells and therefore does not cause liver cell damage like the "Z" mutation does. About 10% of persons that are compound heterozygote for the "Z" and "S" mutations(Pi*ZS) have AAT levels below the protective threshold and are therefore at risk at developing emphysema. All P*ZS are considered to be at increased risk if they smoke.
One mutation in the SERPINA1 gene, is called the "F" mutation. This mutation does not affect the amount of AAT that the liver produces, but is defective. Meaning that these individuals will have normal amounts of serum AAT.
Both infections and high amounts of estrogen (for example pregnancy) can raise the level of serum AAT. It is therefore important to take these two things into consideration when measuring serum AAT.
Main points
- Autosomal Recessive
- Gene: SERPINA1
- Protein: alpha-1 antitrypsin (protease inhibitor, AAT)
- Statistics: carrier frequency 1:3-5000 in scandinavia, 1:5-7000 in US.
-
Most notable mutations:
- Z (can cause liver- and lungdisease)
- S (is generally not considered pathogenic unless it is found in combination with a pathogenic mutation, for example ZS)
- F (normal AAT amount, but non-functional protein. Can cause lungdisease),
- Symptoms: emphysema (mostly >40 years, lower age in smokers. Emphysema presents often in the lung bases), elevated liver enzymes (cirrhosis is rare). Infants can present with liver damage shortly after birth, which can be leathal without liver transplantation.
- Treatment: iv AAT enzyme. Vaccination against lung- and liverinfections. Protect the lungs against smoke and other possibly damaging toxins. Symptomatic treatment.
Cystic Fibrosis
Cystic Fibrosis (CF) is caused by a homozygote or compound heterozygote
pathogenic mutations in the gene CFTR. CFTR is located on the long arm
(q) of chromasome 7. It codes for an ATP-binding cassette transporter
which functions as a chloride channel. It is important in the production
of sweat, saliva, tears, mucus and digestive enzymes. As it moves
chloride through its channel it also controls movement of water which is
necessary in the production of a free flowing mucus. When CFTR does not
function the mucus production becomes thick and accumulates where it is
produced. This can cause blockage of the pancreatic and biliary ducts,
blockage of semen in the vas deferense and blockage of alveoli in the
lungs. The accumulation of mucus in the lungs causes chronic cough,
shortness of breath and bacterial growth leading to frequent lung
infections and Pseudomonas colonization. This in turn damages the
airways, leading to bronchiectasis, and with time respiratory failure,
often requiring lung transplantion. Mucus can also clog the sinuses
leading to sinusitis. In the digestive tract, blockage of pancreatic
ducts can result in indigestion, malnutrition and weight loss, as
pancreatic enzymes cannot reach the duodenum to digest the food we eat.
This accumulation of mucus in the pancreatic ducts can also lead to
inflammation and pancreatitis.
The disease used to be diagnosed by measuring increased chloride in sweat, in a so-called sweat test. Diagnosis is now done via genetic testing, often screening for the most commonly known mutations in the CFTR gene in the region the individual is from. Most countries in the western world, include screening for CF in their newborn screening programs, however it usually does not screen for carrier status. Male carriers of the disease are likelier to have fertility issues compared to non-carriers. This has prompted many fertility centers to screen for CF carrier status among males that seek their help. The most commonly known mutation in the CFTR gene is delta F508 which causes severe symptoms if inherited in homozygous form.
Most men with cystic fibrosis are infertile as the vas deferens atrophies early in embryologic development, this is known as congenital bilateral absence of the vas deferens, CBAVD). Most of them, however, have healthy sperm which can be retrieved via a surgical procedure. It is advised that these men freeze sperm in their younger years, as sperm can become damaged if they have been blocked for a long period of time.
Women with CF can have ovulation issues due to malnutrition and are known to have thicker cervical mucus, dispite this most women with CF are fertile.
The median survival age for children with CF born in recent years is predicted to be about 48-53 years. The leading cause of death is respiratory failure and followed by complications of lung transplantation.

The disease used to be diagnosed by measuring increased chloride in sweat, in a so-called sweat test. Diagnosis is now done via genetic testing, often screening for the most commonly known mutations in the CFTR gene in the region the individual is from. Most countries in the western world, include screening for CF in their newborn screening programs, however it usually does not screen for carrier status. Male carriers of the disease are likelier to have fertility issues compared to non-carriers. This has prompted many fertility centers to screen for CF carrier status among males that seek their help. The most commonly known mutation in the CFTR gene is delta F508 which causes severe symptoms if inherited in homozygous form.
Most men with cystic fibrosis are infertile as the vas deferens atrophies early in embryologic development, this is known as congenital bilateral absence of the vas deferens, CBAVD). Most of them, however, have healthy sperm which can be retrieved via a surgical procedure. It is advised that these men freeze sperm in their younger years, as sperm can become damaged if they have been blocked for a long period of time.
Women with CF can have ovulation issues due to malnutrition and are known to have thicker cervical mucus, dispite this most women with CF are fertile.
The median survival age for children with CF born in recent years is predicted to be about 48-53 years. The leading cause of death is respiratory failure and followed by complications of lung transplantation.
Main points
- Autosomal Rescessive
- Gene: CFTR (cystic fibrosis conductance regulator gene)
- Protein: ATP-binding cassette transporter (Chloride channel)
-
Statistics:
- Carrier frequency: 1:25 (Northern European descent)
- Incidence: 1:2500 (Northern European descent)
- Most notable mutations: deltaF508
- Symptoms: Lung infections and chronic cough leading to bronchiectasis and respiratory failure. Poor growth, weight loss and greasy stool due to undigested fat and proteins. Salty sweat. Infertility in males due to lack of vas deferens. Meconium ileus can be seen in newborns.
- Treatment: Symptomatic treatment. CFTR modulators (mutation specific) can decrease the rate of pulmonary function decline. Lung and Liver transplantation.
Fabry disease
Fabry disease is an X-linked disorder caused by a mutation in the GLA
gene on the X chromosome, which codes for alpha-galactosidase A, a
lysosomal protein important in breaking down globotriaosylceramide
(Gb3). Gb3 is a glycolipid, present in the outer layer of the cell
membrane of different cells. Gb3 degration occurs in the cells
lysosomes, and the first step is for alpha-galactosidase to remove
terminal alpha-galactose residues from Gb3. When the production
alpha-galactosidase A is decreased, it leads to the accumulation of Gb3
in the cells lysosomes, especially in the skin, eyes, heart, brain,
kidney and cells of the peripheral nervous system.
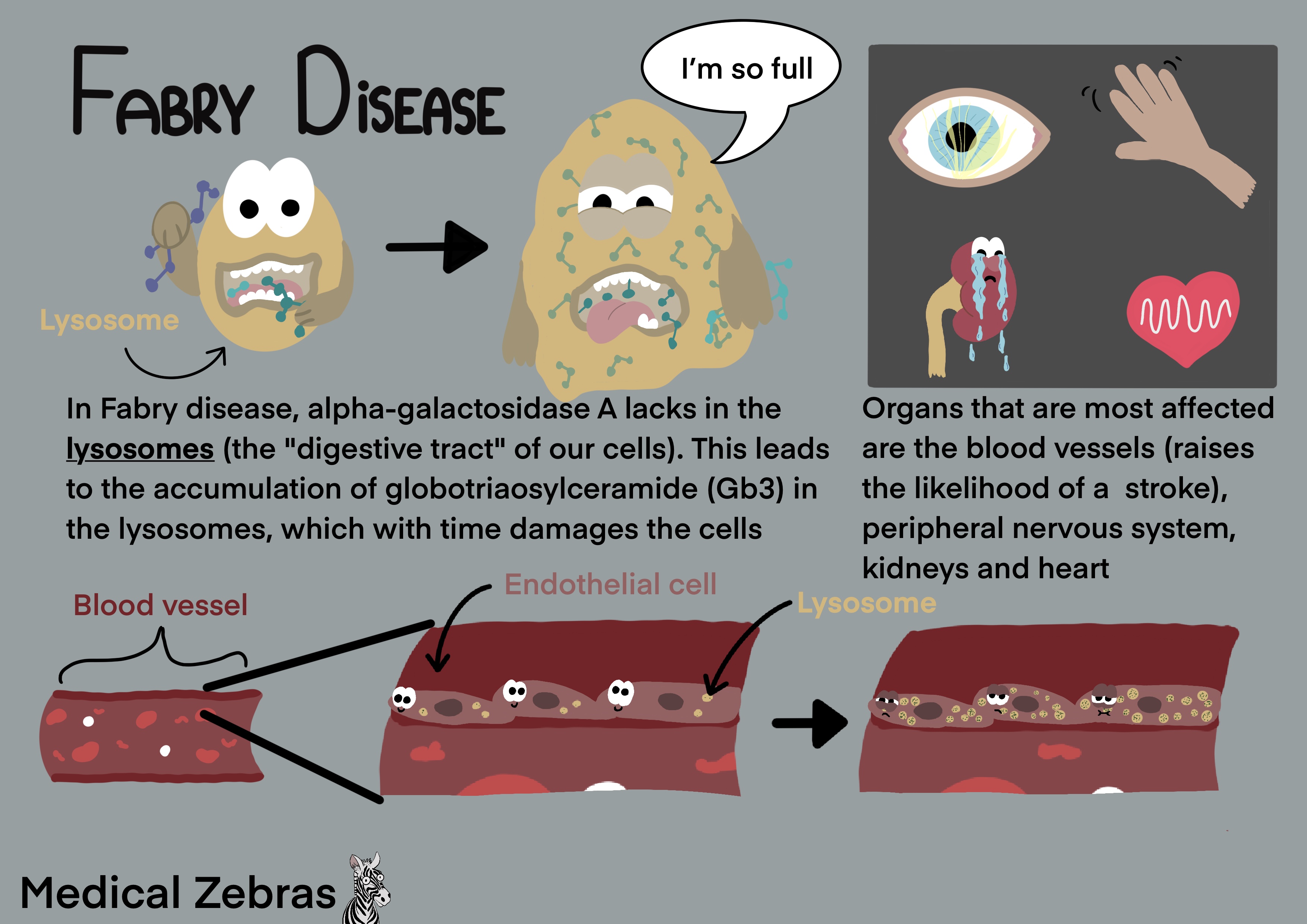
In individuals with enzymic activity of alpha-galactosidase A below 1%, symptoms appear in childhood or adolescense. Whereas if the activity is above 1%, symptom onset can be delayed until after 50 years of age. In younger individuals symptom start is often associated with accumulation of Gb3 in the peripheral nervous system and appears as acroparesthesia. Acroparesthesia is a tingling, burning, pins and needle or pain sensation in the extremities, especially in the fingers or toes, which appears periodically. In the skin the symptoms of Gb3 accumulation can be angiokeratoma and dysfunctional sweating such as lack of sweat (anhidrosis). With time eyesight is affected with the appearance golden-brown or grey opacities with a vortex or whorl pattern in the corneal epithelium known as vortex keratopathy or cornea verticillata. with time kidney and heart function becomes affected as well. End-stage-renal disease can occur from 20 years of age and upward. In the late-onset form, primary manifestastions include decline in heart and kidney function, or stroke.
Due to X-inactivation, women with Fabry disease typically have milder symptoms with later onset. In women symptom severity can however range from asymptomatic to severe symptoms, depending on the X-inactivation pattern.
In individuals with enzymic activity of alpha-galactosidase A below 1%, symptoms appear in childhood or adolescense. Whereas if the activity is above 1%, symptom onset can be delayed until after 50 years of age. In younger individuals symptom start is often associated with accumulation of Gb3 in the peripheral nervous system and appears as acroparesthesia. Acroparesthesia is a tingling, burning, pins and needle or pain sensation in the extremities, especially in the fingers or toes, which appears periodically. In the skin the symptoms of Gb3 accumulation can be angiokeratoma and dysfunctional sweating such as lack of sweat (anhidrosis). With time eyesight is affected with the appearance golden-brown or grey opacities with a vortex or whorl pattern in the corneal epithelium known as vortex keratopathy or cornea verticillata. with time kidney and heart function becomes affected as well. End-stage-renal disease can occur from 20 years of age and upward. In the late-onset form, primary manifestastions include decline in heart and kidney function, or stroke.
Due to X-inactivation, women with Fabry disease typically have milder symptoms with later onset. In women symptom severity can however range from asymptomatic to severe symptoms, depending on the X-inactivation pattern.
Main points
- X-linked
- Gene: GLA
- Protein: Alpha-galactosidase A(lysosomal hydrolase)
- Accumulative substrate: Globotriaosylceramide (Gb3, Ceramide trihexoside)
- Statistics: >1:15.000
- Symptoms: periodic pain with tingling in extremities and neuropathy, heart disease (ventricular hypertrophy, arrhythmia), kidney failure, angiokeratoma, increased risk of stroke, hypohydrosis, cornea verticillata.
-
Treatment:
- Enzyme replacement therapy (agalsidase beta (Fabrazyme) OR agalsidase alfa (Replagal)) given intravenously every two weeks. Some will develope antibodies, and thus will not benefit from the treatment
- chaperone therapy (migalastat, Galafold), given orally once every other day. Binds reversibly to the active site of alpha-galactosidase A and stabilises dysfunctional enzymes.
- There are promising gene therapy results from 2021 (Medin et al, Nature), where CD34+ hematopoietic stem/progenitor cells (HSPCs) were isolated from five men with Fabry disease and infected with a lentivirus containing a healthy alpha-galactosidase A cDNA, and thereafter transplanted into them again. This lead to increased production of alpha-galactosidase A detected by urine and blood samples, but the effect diminished with time.
- Surveillance
Gilbert disease
Gilbert disease is a benign genetic disorder caused by a mutation in the
gene UGT1A1 on chromosome 2 (the same gene as is defective in
Crigler-Najjar syndrome(see below)). The gene codes for an enzyme called
UDP-glucuronosyltransferase, an enzyme responsible for transforming
bilirubin and other lipophilic molecules, into water-soluble
metabolites, by conjugating the molecules with glucuronic acid. As the
molecules become water-soluble they can then be excreted into either the
urine or bile.
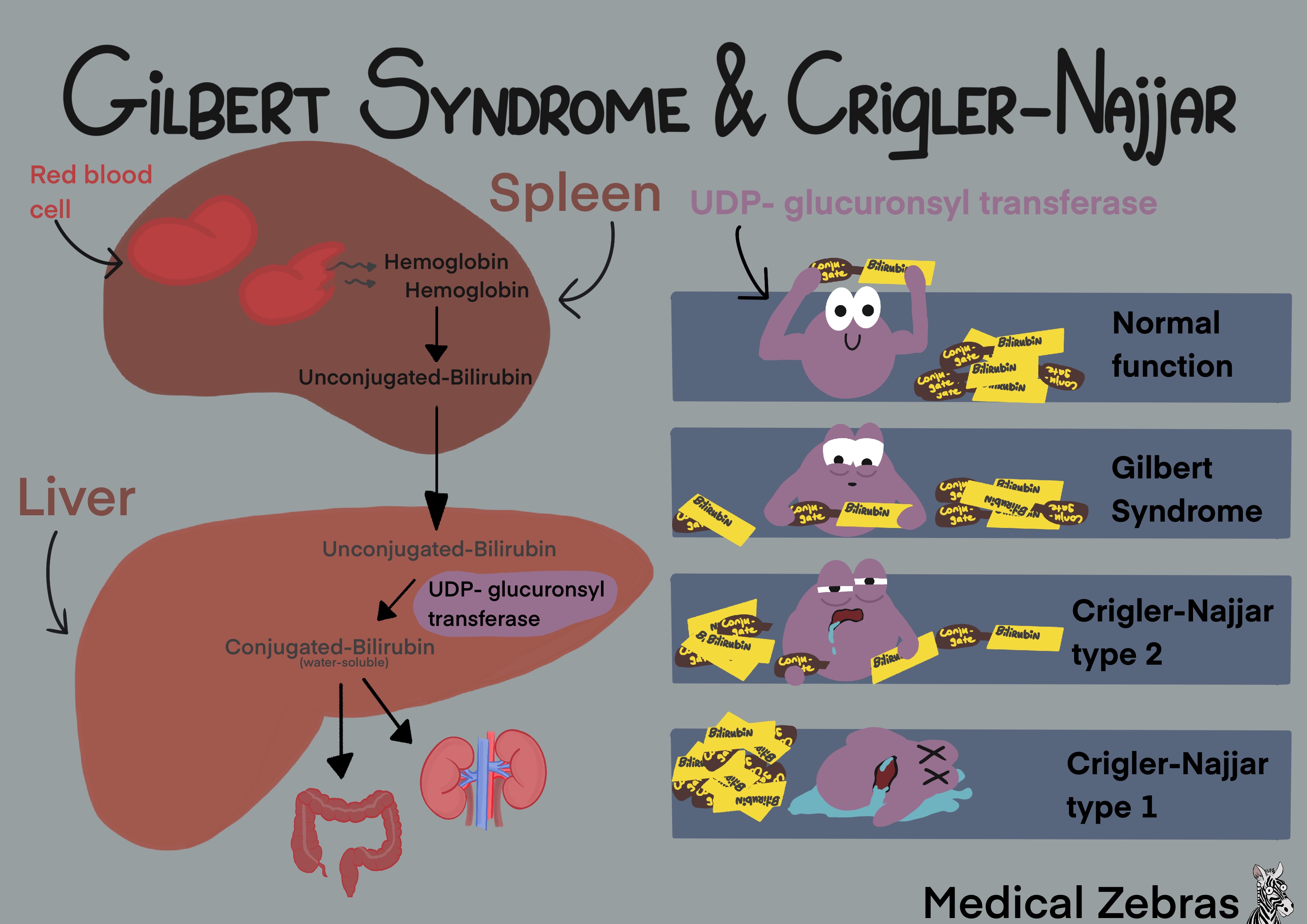
In Gilbert syndrome, UDP-glucuronosyltransferase activity is mildly decreased. This can result in unconjugated-bilirubinemia in response to stress, exercise, dehydration, illness, menstration, fasting or when taking drugs that require the use of UDP-glucuronosyltransferase (for example Atazanavir (HIV medication) and cancer drugs Nilotinib and Irinotecan). Some individuals with Gilbert syndrome might also be at higher risk of statin intolerance, as statins are partly metabolised by UDP-glucuronosyltransferase, especially if combined with Gemfibrozil (VLDL lowering drug), which rises the likelyhood of myopathy. Most individals, however, will never develope jaundice. No other symptoms have been correlated with the syndrome.
The prevalence of Gilbert syndrome is high, 3-7%, and though found on an autosomal chromosome, this condition is more often diagnosed in males than females. This is thought to be due to higher hemoglobin turnover combined with the inhibition of bilirubin glucuronidation by endogenous steroid hormones.
In Gilbert syndrome, UDP-glucuronosyltransferase activity is mildly decreased. This can result in unconjugated-bilirubinemia in response to stress, exercise, dehydration, illness, menstration, fasting or when taking drugs that require the use of UDP-glucuronosyltransferase (for example Atazanavir (HIV medication) and cancer drugs Nilotinib and Irinotecan). Some individuals with Gilbert syndrome might also be at higher risk of statin intolerance, as statins are partly metabolised by UDP-glucuronosyltransferase, especially if combined with Gemfibrozil (VLDL lowering drug), which rises the likelyhood of myopathy. Most individals, however, will never develope jaundice. No other symptoms have been correlated with the syndrome.
The prevalence of Gilbert syndrome is high, 3-7%, and though found on an autosomal chromosome, this condition is more often diagnosed in males than females. This is thought to be due to higher hemoglobin turnover combined with the inhibition of bilirubin glucuronidation by endogenous steroid hormones.
Main points
- Autosomal recessive
- Gene: UGT1A1
- Protein: UDP-glucuronosyltransferase
- Accumulative substrate: unconjugated-bilirubin
- Statistics: prevalence of 3-7% in the general population, up to 10% or higher in Western populations
- Symptoms: mild jaundice develops as result of stress, exercise, dehydration, illness, menstration, fasting or when taking drugs that require the use of UDP-glucuronosyltransferase. Most individuals will never develope symptoms.
- Treatment: the jaundice developed is most often benign, and treatment is not needed
Crigler-Najjar syndrome
Crigler-Najjar syndrome is a genetic disorder caused by a mutation in
the gene UGT1A1 on chromosome 2 (the same gene that is defective in
Gilbert Syndrome (see above)). The gene codes for an enzyme called
UDP-glucuronosyltransferase, an enzyme responsible for transforming
bilirubin and other lipophilic molecules, into water-soluble
metabolites, by conjugating the molecules with glucuronic acid. As the
molecules become water-soluble they can then be excreted into either the
urine or bile. Unlike in Gilbert Syndrome, here the enzyme is either
absent (type I) or severly defective (type II).
In type I there is no bilirubin conjugation leading to sky-high plasma bilirubin, bilirubin encephalopathy (kernicterus) and death in infancy if untreated. Bilirubin encephalopathy in infants causes lethargy, hypotonia, brain damage as well as other symptoms.
In type II there is some conjugation of bilirubin and lifespan is usually normal and kernicterus rare. As there is some UDP-glucuronosyltransferase activity, type II can be treated with phenobarbital which increases liver enzyme synthesis and lifespan is usually normal.
In type I there is no bilirubin conjugation leading to sky-high plasma bilirubin, bilirubin encephalopathy (kernicterus) and death in infancy if untreated. Bilirubin encephalopathy in infants causes lethargy, hypotonia, brain damage as well as other symptoms.
In type II there is some conjugation of bilirubin and lifespan is usually normal and kernicterus rare. As there is some UDP-glucuronosyltransferase activity, type II can be treated with phenobarbital which increases liver enzyme synthesis and lifespan is usually normal.
Main points
- Autosomal recessive
- Gene: UGT1A1
- Protein: UDP-glucuronosyltransferase
- Accumulative substrate: unconjugated-bilirubin
- Statistics: 1-9:100.000
-
Symptoms:
- Type I: Kernicterus and death in infancy if untreated.
- Type II: Persistant jaudice, kernicterus is rare, but the likelihood of it is increased with illness or fasting.
-
Treatment:
- Type I: Liver transplatation and phototherapy.
- Type II: phenobarbital.
Glucose-6-phosphate dehydrogenase deficiency (favism)
Glucose-6-phosphate dehydrogenase deficiency is caused by a mutation in
the gene G6PD, which codes for glucose-6-phosphate dehydrogenase (G6PD).
G6PD is a cytosolic enzyme important in the pentose phosphate pathway
which is an especially important metabolic pathway in red blood cells.
G6PD reduces NADP+ to NADPH by simutaneously oxidizing
glucose-6-phosphate. NADPH is then used to maintain the levels of
glutathione, which is important in protecting the cell from damage from,
for example, reactive oxygen species (ROS), free radicals and more. That
is why when the G6PD enzyme is defective, it leaves the cell vulnerable
to ROS and free radicals which increase in response to stresses such as
infection, certain medications or diet (i.e. favabeans). These
situations can therefore cause hemolysis in the red blood cells in
individuals with G6PD deficiency, which can lead to hemolytic anemia and
splenomegaly due to sequestration of red blood cells. Additionally,
infants with G6PD are at increased risk of neonatal jaundice.

Main points
- X-linked
- Gene: G6PD
- Protein: Glucose-6-phosphate dehydrogenase
- Statistics: Most common human enzyme deficiency. More common in individuals of African, Mediterranian and Asian decent.
- Symptoms: Hemolytic crisis in response to infections, certain foods and medications.
- Treatment: Avoidance of agents that can invoke hemolytic episodes. Transfusion if needed.
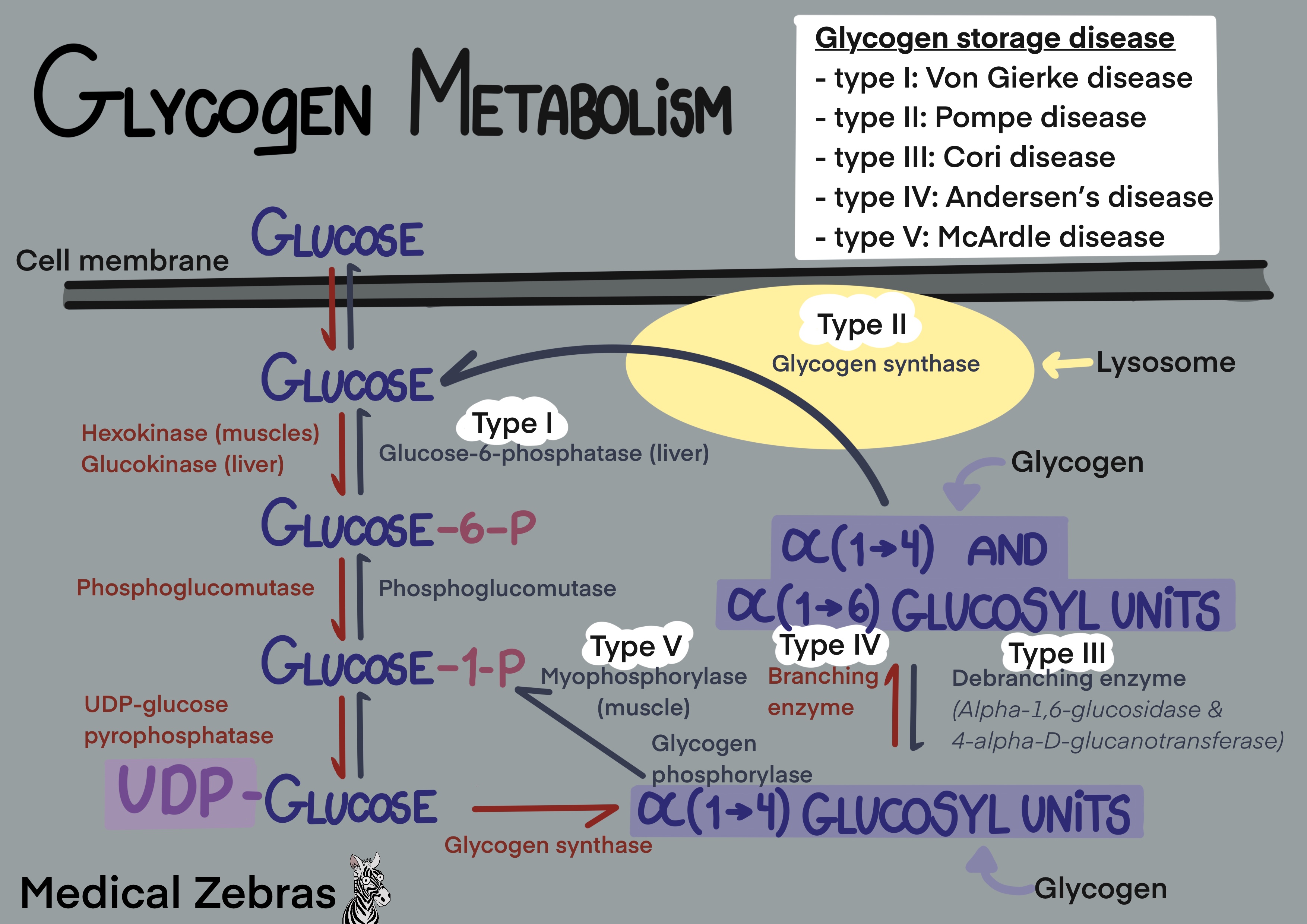
Glycogen storage disease type I - von Gierke disease
Glycogen storage disease type I is caused by either a mutation in the
gene G6PC1 (type Ia) or SLC37A4 (type Ib). Both genes code for proteins
vital to the regulation of blood glucose levels.
Type Ia: is caused by a mutation in G6PC1 that codes for glucose-6-phosphatase, and is responsible for aproximately 80% of cases of Glycogen storage disease type I. This gene codes for glucose-6-phosphatase which mediates the last step of both gluconeogenesis (transforming non-carbohydrate substrates into glucose) and glycogenolysis (the breakdown of glycogen into glucose), where it removes a charged phosphate group from Glucose-6-phosphate, which then becomes usable glucose. The main reason glucose is phosphorylated is to prevent its outflow out of the cell. Thus if the cells are unable to remove this phosphate from the glucose, it becomes trapped in the cells and the body is unable to access it when extra glucose is needed, for example during fasting. As glygogen (a stored form of glucose) is stored in the liver, a defect in glucose-6-phosphatase leads to an enlarged liver and extreme hypoglycemia during fasting. As the body utilises other methods of gaining energy during the crisis, both protein and fats are broken down which leads to both hyperuricemia and hyperlipidemia. There can also be a buildup of lactic acid.
Most infants first present with symptoms at 3-4 months of age, as that is the time when infants start to sleep longer, requiring the body to be able to use glycogen storage to raise the blood glucose during the sleep fasting. As affected infants lack a functioning glucose-6-phosphatase they will develope symptoms of hypoglycemia, such as irritibility, lethargy, breathing problems and seizures.
As individuals get older, they tend to have thin extremities and short stature. Hyperlipidemia can with time cause deposits of cholestrol in the skin (xanthomas) and hyperuricemia can lead to gout. Adenomas can develop in the liver. As glucose-6-phosphatase is also prevalent in the kidneys, the kidneys become enlarged.
Type Ib: is caused by a mutation in SLC37A4 that codes for glucose-6-phosphate translocase which is responsible for translocating glucose-6-phosphate from the cytoplasm to the lumen of endoplasmic reticulum where glucose-6-phosphatase can remove the phosphate from the glucose. When the enzyme is dysfunctional, glucose-6-phosphate cannot reach glucose-6-phosphatase and thus becomes stuck in the cell, like in glucogen storage disease type Ia.
Symptoms mirror what is seen in type Ia. These individuals also have an increased risk for developing neutropenia, often apparent in the first few years of life. This increases the likelihood for recurrent bacterial infections.
Type Ia: is caused by a mutation in G6PC1 that codes for glucose-6-phosphatase, and is responsible for aproximately 80% of cases of Glycogen storage disease type I. This gene codes for glucose-6-phosphatase which mediates the last step of both gluconeogenesis (transforming non-carbohydrate substrates into glucose) and glycogenolysis (the breakdown of glycogen into glucose), where it removes a charged phosphate group from Glucose-6-phosphate, which then becomes usable glucose. The main reason glucose is phosphorylated is to prevent its outflow out of the cell. Thus if the cells are unable to remove this phosphate from the glucose, it becomes trapped in the cells and the body is unable to access it when extra glucose is needed, for example during fasting. As glygogen (a stored form of glucose) is stored in the liver, a defect in glucose-6-phosphatase leads to an enlarged liver and extreme hypoglycemia during fasting. As the body utilises other methods of gaining energy during the crisis, both protein and fats are broken down which leads to both hyperuricemia and hyperlipidemia. There can also be a buildup of lactic acid.
Most infants first present with symptoms at 3-4 months of age, as that is the time when infants start to sleep longer, requiring the body to be able to use glycogen storage to raise the blood glucose during the sleep fasting. As affected infants lack a functioning glucose-6-phosphatase they will develope symptoms of hypoglycemia, such as irritibility, lethargy, breathing problems and seizures.
As individuals get older, they tend to have thin extremities and short stature. Hyperlipidemia can with time cause deposits of cholestrol in the skin (xanthomas) and hyperuricemia can lead to gout. Adenomas can develop in the liver. As glucose-6-phosphatase is also prevalent in the kidneys, the kidneys become enlarged.
Type Ib: is caused by a mutation in SLC37A4 that codes for glucose-6-phosphate translocase which is responsible for translocating glucose-6-phosphate from the cytoplasm to the lumen of endoplasmic reticulum where glucose-6-phosphatase can remove the phosphate from the glucose. When the enzyme is dysfunctional, glucose-6-phosphate cannot reach glucose-6-phosphatase and thus becomes stuck in the cell, like in glucogen storage disease type Ia.
Symptoms mirror what is seen in type Ia. These individuals also have an increased risk for developing neutropenia, often apparent in the first few years of life. This increases the likelihood for recurrent bacterial infections.
Main points
- Autosomal recessive
- Gene: G6PC1 or SLC37A4
- Protein: glucose-6-phosphatase or glucose-6-phosphate translocase
- Statistics: 1:100.000 overall. Prevalence 5 times higher in the Ashkenazi Jewish population. (80% are type Ia and 20% are type Ib).
- Symptoms: hepatomegaly, hypoglycemia, hyperlipidemia, hyperuricemia, increased blood lactate. Increased risk of neutropenia in type Ib.
- Treatment: Stict dietary changes and frequent eating to prevent hypoglycemia. Lipid lowering medications to resolve hyperlipidemia, angiotensin-converting enzyme to treat microalbuminuria, allopurinol or citrate supplement to treat hyperuricemia. Liver and kidney transplatation if needed. Tight surveillance of possible symptoms.
Glycogen storage disease type II - Pompe disease
Glycogen is metabolised both in the cytosol and lysosome. The enzyme
responsible for glycogen degradation in the lysosomes is lysosomal alpha
glucosidase. Lysosomal alpha glucosidase is coded by the gene GAA, and
its function is to separate the alpha-1,4 and alpha-1,6-glycosidic bonds
of glycogen in the lysosome. In glycogen storage disease type II or
Pompe disease, the gene GAA is mutated, rendering the lysosomal alpha
glucosidase dysfunctional. This leads to the accumulation of lysosomal
glycogen everywhere in the body, where the cardiac and skeletal muscles
cells are most severly affected. Symptoms typically present either in
infancy (early-onset) or adulthood (late-onset) which correlates with
the degree enzyme deficiency.
Early-onset: Cardinal symptoms of the classic form are cardiomegaly, hypertrophic cardiomyopathy and hypotonia presenting in the first months of life. Other symptoms that can follow are feeding difficulties leading to poor weight gain and respiratory difficulties. This condition most often leads to death of the individual by 2 years of age due to heart failure. Those who have non-classic symptoms present by age 1. They may have cardiomyopathy, but without heart failure. Most apparent symptoms are progressive muscle weaknesss that delays motor development and affects breathing, often leading to the death of the individual in early childhood.
Late-onset: Symptoms appear at any time during early childhood as progressive muscle weakness, primerily in the legs and torso, affecting breathing muscles. The heart is usually not affected.
Early-onset: Cardinal symptoms of the classic form are cardiomegaly, hypertrophic cardiomyopathy and hypotonia presenting in the first months of life. Other symptoms that can follow are feeding difficulties leading to poor weight gain and respiratory difficulties. This condition most often leads to death of the individual by 2 years of age due to heart failure. Those who have non-classic symptoms present by age 1. They may have cardiomyopathy, but without heart failure. Most apparent symptoms are progressive muscle weaknesss that delays motor development and affects breathing, often leading to the death of the individual in early childhood.
Late-onset: Symptoms appear at any time during early childhood as progressive muscle weakness, primerily in the legs and torso, affecting breathing muscles. The heart is usually not affected.
Main points
- Autosomal recessive
- Gene: GAA
- Protein: lysosomal alpha glucosidase
- Statistics: 1:40.000 worldwide
- Symptoms: Cardiomyopathy, hypotonia and respiratory distress in the early-onset form. Progressive muscle weakness affecting respiration in late-onset form, where the heart is usually spared.
- Treatment: Enzyme replacement therapy (some will develope antibodies, and will therefore not benefit from this treatment). Dietary changes. Otherwise symptomatic. Surveillance.
Glycogen storage disease type III - Cori disease
Glycogen storage disease type III is caused by a mutation in the gene AGL, which codes for a glycogen debrancher enzyme, which has two catalytic functions both important in breaking down glycogen.Type III is divided into two subcategories depending on if both the liver and muscles are affected, called type IIIa (85% of cases), or just the liver, called type IIIb (15% of cases).
Symptoms appear in infancy or early childhood with hepatomegaly and elevated liver ezymes. As glycogen breakdown is affected, the availability of blood glucose during fasting is limited and fat is metabolised, leading to hyperlipidemia and ketotic hypoglycemia. This with time affects the growth of the individual and can therefore lead to short stature. Muscle weakness can become apparent later in life and can extend to the heart, increasing the risk for cardiomyopathy and cardiac hypertrophy.
Note that uric acid and lactate are usually normal, unlike in Von Gierke (glycogen storage disease type I).
Main points
- Autosomal recessive
- Gene: AGL
- Protein: Glycogen debrancher enzyme
- Statistics: 1:100.000 (Europe)
- Symptoms: hepatomegaly, elevated liver enzymes, fasting hypoglycemia and hyperlipidemia in infancy or early childhood. Later skeletal and cardiac muscles are affected in type IIIa.
- Treatment: dietary changes and avoidance of fasting. Liver transplantation only if severe cirrhosis or liver cancer. Surveillance.
Glycogen storage disease type IV - Andersen's disease
Glycogen storage disease type IV is caused by a mutation in the GBE1
enzyme that codes for glycogen branching enzyme. The glycogen branching
enzyme is responsible for enabling the transfer of alpha-1,4 glycosyl
units from the outer end of a glycogen molecule to an alpha-1,6
position, thereby changing glycogen from a linear structure to a
branched structure which increases the solubility of glycogen. When the
glycogen branching enzyme is defective, it leads to the accumulation of
abnormally structured glycogen which impairs the function of the liver,
muscles and other organs. Muscles are most severly affected, with
symptoms often appearing as hypotonia and muscle atrophy, which can lead
to respiratory distress. Dilated cardiomyopathy can develope as well as
hepatomegaly that can in some cases lead to cirrhosis.
The symptoms vary from appearing perinatally to up to the second decade of life. Symptoms tend to be more severe the sooner they appear.
Individuals who show symptoms perinatally or shortly after birth, usually die in the neonatal period or in infancy.
The symptoms vary from appearing perinatally to up to the second decade of life. Symptoms tend to be more severe the sooner they appear.
Individuals who show symptoms perinatally or shortly after birth, usually die in the neonatal period or in infancy.
Main points
- Autosomal recessive
- Gene: GBE1
- Protein: glycogen branching enzyme
- Statistics: 1:600.000-800,000 overall
- Symptoms: Hypotonia, respiratory distress, dilated cardiomyopathy, liver dysfunction and hepatomegaly. Symptoms can appear perinatally to early adulthood, where symptoms are more severe the sooner they appear.
- Treatment: Symptomatic and tight surveillance.
Glycogen storage disease type V - McArdle disease
Glycogen storage disease type V is caused by a mutation in the gene
PYGM, which codes for myophosphorylase, a glycogen phosphorylase located
in the muscles. Myophosphorylase is responsible for breaking down
glycogen into glucose-1-phosphate in the muscles, a vital step in
glycogen breakdown. Symptoms appear during exercise when the muscles are
in the need of breaking down glycogen to access more glucose. As the
muscles are unable to release glucose from glycogen due to a defect in
myophosphorylase, energy is depleted. This leads to cramping, muscle
pain and fatigue. If exercise is continued without rest this damages the
muscle cells as they are unable to supply energy, leading to
rhabdomyolysis. As muscle cells are damaged, myoglobin leaks out of the
cells causing myoglobinuria (red urine). Exercise can usually continue
with little or no discomfort after a brief rest, this phenomenon is
called "the second-wind phenomenon" and is due to increased muscular
blood flow.
Symptoms can start anywhere from infancy to adulthood, but most individuals start experiencing symptoms in their teenage years and up.
Symptoms can start anywhere from infancy to adulthood, but most individuals start experiencing symptoms in their teenage years and up.
Main points
- Autosomal recessive
- Gene: PYGM
- Protein: Myophosphorylase
- Statistics: Unkown.
- Symptoms: Exercise intolerance (muscle cramps and pain, fatigue). Myoglobinurea if exercise is continued without rest. After rest exercise can continue (second-wind phenomenon).
- Treatment: Avoidance of strenuous exercise. Dietary changes.
McCune Albright Syndrome
McCune Albright Syndrome is caused by an early embryonic, activating or
gain-of-function,21° postzygotic mutation in the gene GNAS, which
is located on the long arm of chromosome 20.
Main points
- Autosomal dominant, postzygotic mutation (mosaic state)
- Gene: GNAS
- Protein: Guanine Nucleotide Binding Protein (G Protein), Alpha
- Statistics: sporatic
- Symptoms:
- Treatment: