Chromosome Analysis (Karyotyping)
To visualize the chromosomes we can utilize the fact that during the
prometaphase (early metaphase) of cell division, all of the chromosomes
have condensed in preparation for cell division. By applying various
chemicals (for example colchicine) to cells, they can be arrested in
prometaphase allowing for analysis. Once in prometaphase the chromosomes
can then be visualized by staining the cells with Giemsa stain. This
staining reveals distinctive light and dark colored bands in the
chromosomes. Dark bands are more condensed areas in the chromosome, in
which more of the stain binds to the DNA. These areas are also called
Giemsa positive or G-positive. The lighter bands are therefore areas of
the chromosome where less Giemsa stain is bound and are Giemsa
(G-)negative. These bands are then identifiable under a microscope.
G-positive bands are also referred to as heterochromatic regions, they
contain fewer genes and the DNA is less transcriptionally active as the
tightly bound DNA leaves less space for the transcription machinery. In
comparison, G-negative bands or euchromatin regions are more loosely
bound and are more transcriptionally active. By examining the length of
the chromosomes, the number of G-bands and the placement of the
centromere - the area where two sister chromatids link together during
cell division - each chromosome can be identified. Normal karyotyping
can therefore only identify if chromosomes are missing, whether extra
chromosomes are present or if large translocations or deletions have
occurred. High-resolution banding, on the other hand, can identify up to
550 to 800 bands. This means that one can identify more subtle
structural abnormalities such as deletions, duplications, inversions and
translocations, as long as the changes involve segments larger than 5-10
Megabasepairs. If enough cells are examined, mosaicism can sometimes be
detected.

Pros: Great for detecting trisomy and the detection
of whole chromosome deletions.
Cons: Can only detect very large changes in the chromosome structure and is a laborious process as the synchronization of the cells (i.e. arresting as many cells as possible in prometaphase) before staining is time consuming. Examples of uses in the field
Examples of uses in the field
~ Trisomy detection, trisomy 21 translocation detection
~ Sex determination
~ Detection of ring chromosomes
Cons: Can only detect very large changes in the chromosome structure and is a laborious process as the synchronization of the cells (i.e. arresting as many cells as possible in prometaphase) before staining is time consuming. Examples of uses in the field
Examples of uses in the field
~ Trisomy detection, trisomy 21 translocation detection
~ Sex determination
~ Detection of ring chromosomes
Fluorescence in situ hybridization (FISH)
Fluorescence in situ hybridization is similar to karyotyping in that cells are also fixed in prometaphase or metaphase. Thereafter fluorescently labeled probes are used to identify areas of interest. A couple of different probes can be designed to identify different areas of interest simutaniously. This method gives a resolution of a few kilobases.
Pros: Great for screening for common large
translocations such as can occur in many hematological cancers, e.g.
when looking for the philadelphian chromosome.
Cons: low resolution and you have to know exactly what you are looking for, so you can design the right probes to use.
Examples of uses in the field
~ to detect commonly known translocations in hemotological cancers. For example the 9:22 translocation associated with chronic myeloid leukemia (CML).
~ Detecting the chromosome deletion (15q11.2-13 deletion) associated with 60-70% of cases of Angelman syndrome.
Cons: low resolution and you have to know exactly what you are looking for, so you can design the right probes to use.
Examples of uses in the field
~ to detect commonly known translocations in hemotological cancers. For example the 9:22 translocation associated with chronic myeloid leukemia (CML).
~ Detecting the chromosome deletion (15q11.2-13 deletion) associated with 60-70% of cases of Angelman syndrome.

DNA Microarrays (CGH- and SNP-Arrays)
DNA Microarrays are chips, sometimes called biochips, covered with
microscopic DNA that corresponds to certain DNA sequences. Microarrays
can be used to measure expression levels of genes as well as to
genotype. Each chip is covered with a few hundred thousand to millions
of probes, each consisting of thousands of the same single DNA sequence
from representative sequences across the genome of interest. Each type
of probe is situated in their own dedicated spot on the chip,
corresponding to a certain area of the genome. There are two different
types of microarrays that are mostly used today; CGH(comparative genomic
hybridization)-array or SNP(single nucleotide polymorphism)-array.
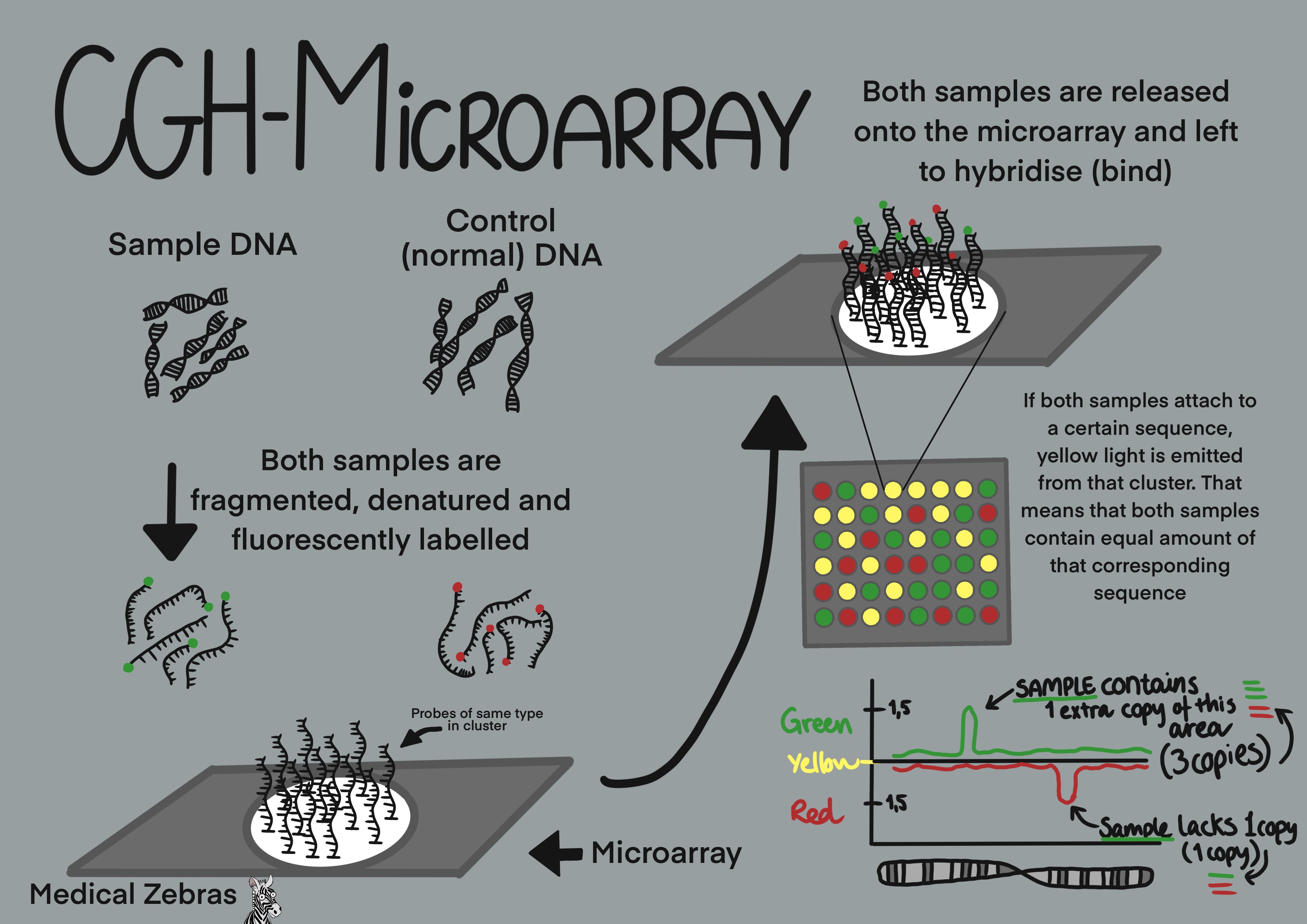
CGH-arrays are used to analyse copy number variations (CNVs) in the
genome. Here the test DNA is fluorescently labelled as well as control
DNA (normal DNA), both in its own color after it has been fragmented.
Both samples are then released onto the chip and let to hybridize (bind)
to the DNA strands on the chip itself. If the corresponding DNA sequence
is only present on the test sample, the particular spot on the chip will
emit only the color of the test sample when excited by laser. If the
corresponding DNA sequence only corresponds to a sequence on the control
DNA, that particular spot will only emit the control DNA color when
excited by laser. If both the sample and control DNA can hybridize to
the chip DNA sequence, the colors blend when that spot on the chip is
excited. This way the quantity of each DNA segment of the test DNA
sample can be determined.
SNP-Arrays, however, use DNA probes from regions in the genome that are known to show differences between individuals at a single base pair site. Because of this, the SNP-arrays can detect consanguinity, limited outbreeding, triploidy and uniparental disomy (UPD) as well as copy number variance. The SNP arrays can sometimes detect mosaicism if the color ratio does not fit a simple deletion or duplication (though not low-levels of mosaicism). SNPs arrays only require test DNA, where it is first fragmented and later fluorescently labelled. The results are then compared to a reference set that has been determined from many other previously run normal DNA samples.
The resolution of DNA microarrays varies between laboratories, and can range from 50kb - 400kb, some with lower resolution, as it depends on how many probes the microarray contains.
SNP-Arrays, however, use DNA probes from regions in the genome that are known to show differences between individuals at a single base pair site. Because of this, the SNP-arrays can detect consanguinity, limited outbreeding, triploidy and uniparental disomy (UPD) as well as copy number variance. The SNP arrays can sometimes detect mosaicism if the color ratio does not fit a simple deletion or duplication (though not low-levels of mosaicism). SNPs arrays only require test DNA, where it is first fragmented and later fluorescently labelled. The results are then compared to a reference set that has been determined from many other previously run normal DNA samples.
The resolution of DNA microarrays varies between laboratories, and can range from 50kb - 400kb, some with lower resolution, as it depends on how many probes the microarray contains.
Pros: Easy method to detect CNVs (microduplications
and microdeletions).
Cons: Only detects the DNA sequence that the array has defined, it is therefore not 100% genomic coverage. Cannot detect balanced genomic abnormalities, like balanced translocation or inversions (because the DNA is fragmented). Cannot detect ring chromosomes. CGH-arrays cannot detect triploidy. SNP-arrays cannot detect low-level mosaicism.
Examples of uses in the field
~ Screening for genetic abnormalities in children with mental retardation of unknown cause.
~ Prenatal screening for genetic abnormalities after ultrasound abnormalities have been detected.
Cons: Only detects the DNA sequence that the array has defined, it is therefore not 100% genomic coverage. Cannot detect balanced genomic abnormalities, like balanced translocation or inversions (because the DNA is fragmented). Cannot detect ring chromosomes. CGH-arrays cannot detect triploidy. SNP-arrays cannot detect low-level mosaicism.
Examples of uses in the field
~ Screening for genetic abnormalities in children with mental retardation of unknown cause.
~ Prenatal screening for genetic abnormalities after ultrasound abnormalities have been detected.
Polymerase-chain-reaction (PCR)
PCR is used to amplify a small portion of DNA with the help of primers
(specially designed single stranded DNA sequences that bind on either
side of a region of interest, i.e. the DNA region that we want to
study). Most often primers are ca. 20 nucleotides in length and are
designed to amplify DNA regions about 100-200 nucleotides long. It is
important that the primers are not too small, as it is then likelier
that they become more nonspecific to the region of interest (meaning
that they can bind to other places on the DNA strand, not just to the
region of interest).
PCR can be used to amplify tiny amounts of DNA, and is often used to prepare samples for further DNA analysis.
PCR can be used to amplify tiny amounts of DNA, and is often used to prepare samples for further DNA analysis.
The PCR mix
- DNA of interest (often reffered to as the DNA template): Your sample.
-
Primers: Specially designed short single strands of
DNA. One is forward, meaning that it can attach to the reverse lying
DNA strand and one reverse that can attach to the forward lying DNA
strand. Primers are made of oligonucleotides, often 20 nucleotides
in length each and are designed to attach on either side of the
region of interest (the area you want to amplify for study). As you
can only study one region of interest for each pair of primers,
multiple pairs of primers must be used to investigate more than one
region at a time. This is called multiplex PCR when working with
many pairs of primers at one time.
If you want to do a more generalized study of the whole genome, so-called random primers can be used. In this case you use primers designed to be able to attach to the DNA in various regions of the genome with the hopes of replicating the whole of the genome and not just a tiny part of it as is the case with traditional PCR. - DNA polymerase: An enzyme that with the help of a cofactor (see below), elongates DNA by attaching to an attached primer. The most commonly used is Taq polymerase which is heat resistant which prevents its degredation in the PCR process.
- dNTP: nucleotides (A, T, C, G). Building blocks for the polymerase chain reaction, which will become our future amplified region of interest.
- Buffer solution: A ionic fluid used to create the optimal environment for the polymerase to work in.
- Bivalent Cations: A cofactor for the polymerase enzyme. Most often magnesium (Mg+2) or manganese (Mn+2). Enables the incorparation of dNTP during polymerization.
The PCR process (one cycle)
- Denaturing or DNA melting: The sample solution is heated to 94-95°C. This causes the breaking of the hydrogen bonds in the double stranded DNA, making single stranded DNA.
- Annealing: Here the sample tempurature is lowered to 50-56°C at this temperature primers can attach to the single stranded DNA. The precise temperature depends on the primer melting tempurature (Tm) of the primers that are being used.
- Extending: Finally the sample is heated to 72-80°C, which is the optimal temperature for the DNA polymerase to work.
Gel Electrophoresis
The classic method of DNA analysis. Here we make an agarose gel. Wells
are made in the gel for our samples and then an electric current is
passed through the gel for a certain amount of time to move the DNA,
which is negatively charged, from the negative pole to the positive
pole. During this process smaller fragments move faster through the gel
than larger fragments and thus we get a separation of the fragments
within the sample. Then we can take a photo of the gel (often under UV
light) and compare the length traveled by the sample DNA with a DNA
ladder which contains DNA fragments of various known sizes.
Pros: Great for genotyping, when you know the exact
mutation you are looking for.
Cons: You have to know what you are looking for and design special primers for each specific mutation. A positive control is needed to be sure that the primer works.
Examples of uses in the field
~ to differentiate between a mutant mouse and a wild type one (genotyping).
Cons: You have to know what you are looking for and design special primers for each specific mutation. A positive control is needed to be sure that the primer works.
Examples of uses in the field
~ to differentiate between a mutant mouse and a wild type one (genotyping).

Sanger-Sequencing
~ Fluorescent dye-termination sequencing
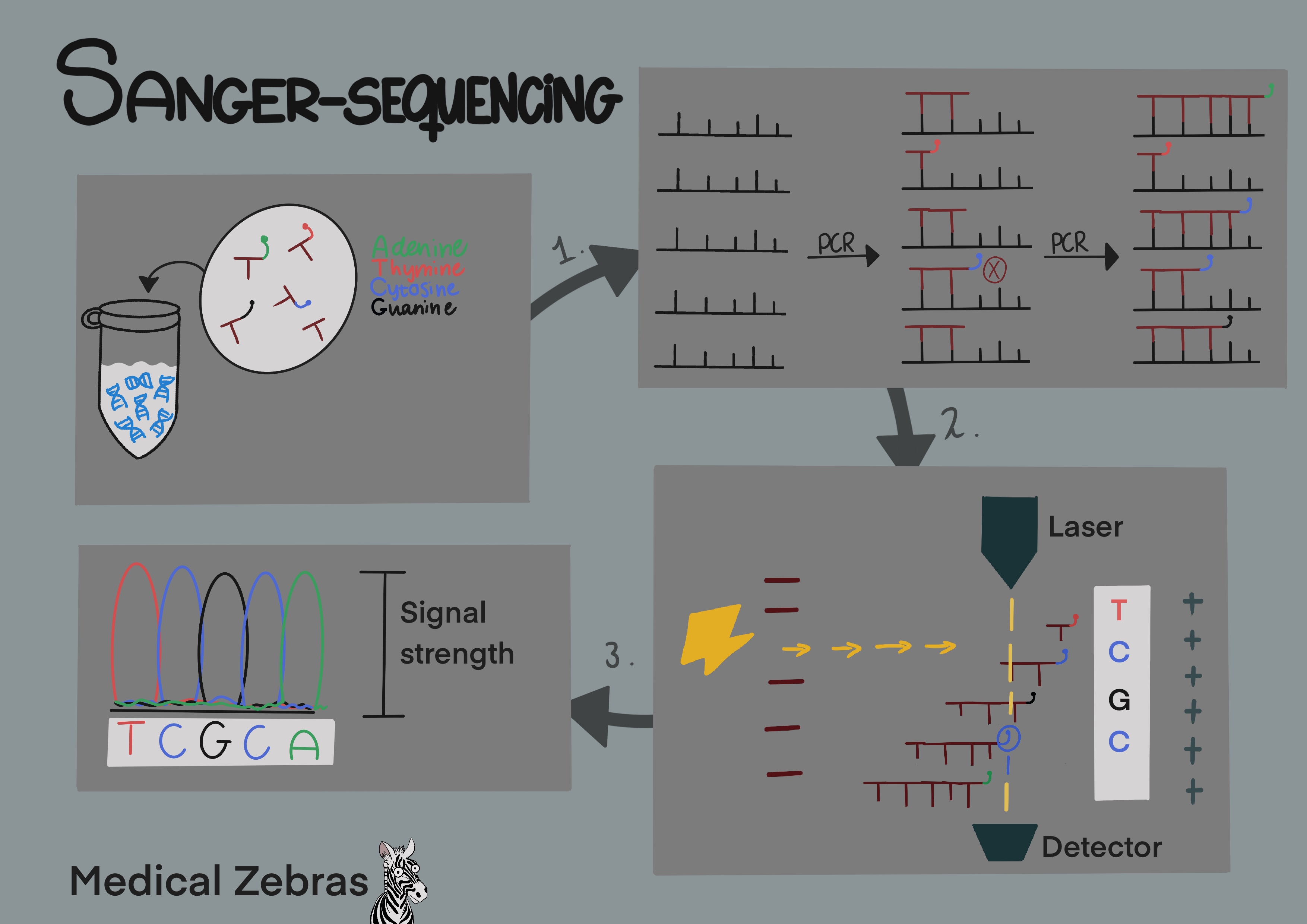
After a PCR cycle, the targeted DNA has been amplified. In Sanger
sequencing the next step is to add more nucleotides to the mix with some
of them being fluorescently marked with each base with its own color (A
- green, T - red, C - blue, G - black) as well as a reverse or forward
primer (depending on which strand we wish to examine). By running this
new mixture through additional PCR thermocycles, further amplification
occurs, however, whenever a flourescently marked nucleotide binds to a
strand the flourescent marker prevents a new nucleotide from binding to
that strand stopping the reaction, thus determining the strands length.
After enough amplification we end up with a mixture where a
flourescently marked nucleotide is sitting in each potential spot of our
target DNA strand. This new mixture can then be transfered through
capillary gel with an electric current, in a process similar to gel
electrophoresis. In this situation there is a lazer waiting to detect
the fluorescent basepairs as they pass through the gel. As smaller DNA
travels the fastest, it will be the first one to be detected by the
lazer with each subsequent length of strand following until the largest
strand is the last to be detected. When we run the data through Sanger
sequencing software, it tells us what kind of fluorescent signal was
detected first, which would correspond to the first base of that DNA
string. The second signal would be the second base etc.
Pros: cheap and relatively fast compared to
whole-genome sequencing (WGS) and whole-exome-sequencing (WES).
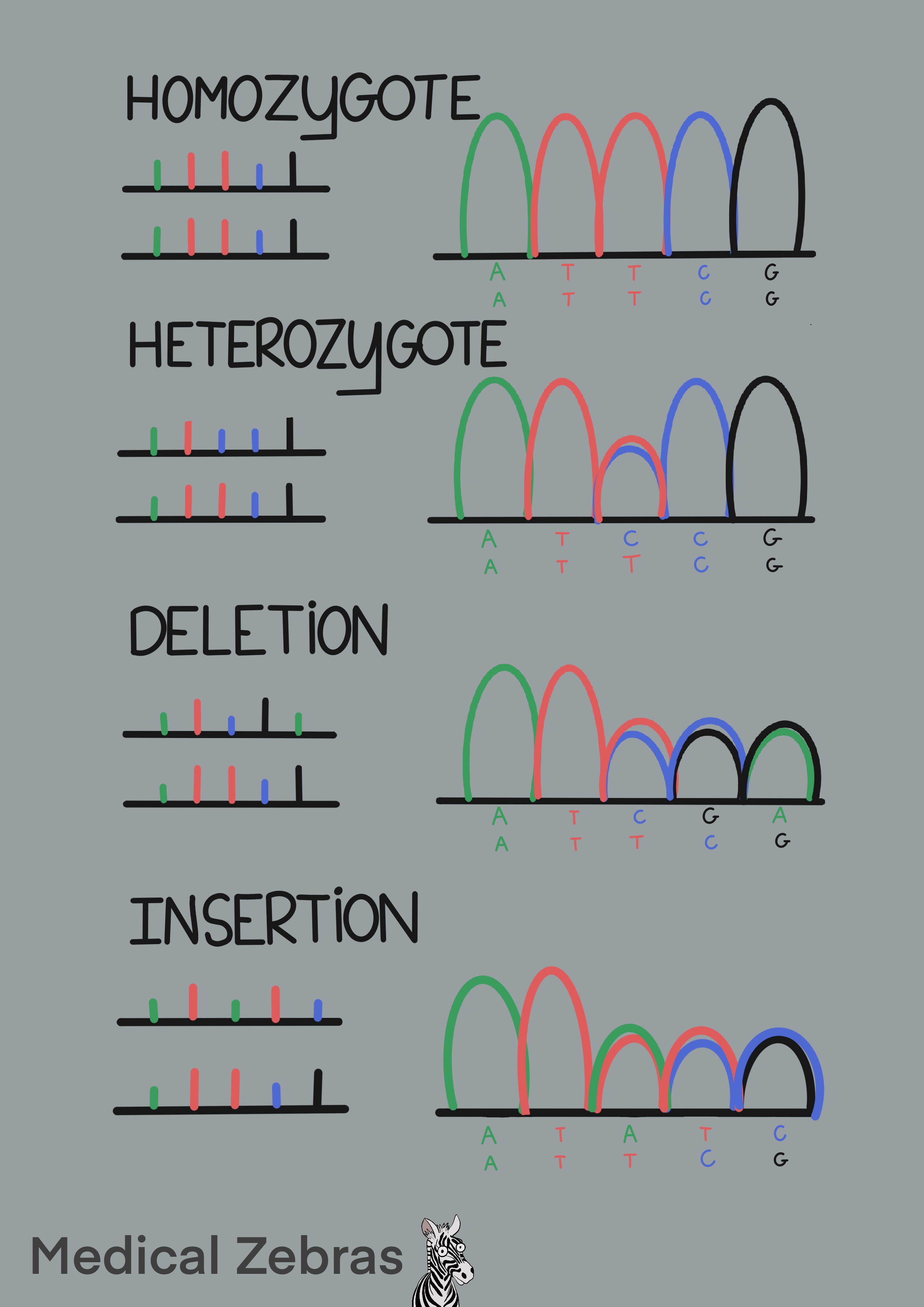
Cons: Only one gene/site can be checked in each run. Hard to detect large deletions if the person is heterozygote (then the normal strand will be amplified and that could mask the large deletion).
Examples of uses in the field
~ check carrier status in families with known familial mutations.
Cons: Only one gene/site can be checked in each run. Hard to detect large deletions if the person is heterozygote (then the normal strand will be amplified and that could mask the large deletion).
Examples of uses in the field
~ check carrier status in families with known familial mutations.
Whole-exome-sequencing (WES)
Whole exome sequencing is a technique where all known exons (protein
coding DNA strips) in a genome are sequenced. As most genetic diesases
known today are diseases with defects in the exons, this method has high
yield of catching relevant variants, compared to
whole-genome-sequencing. Todays main Next Generation Sequencing
platforms (NGS) are Ion Torrent and Illumina platfoms (MiSeq, HiSeq,
NovaSeq).
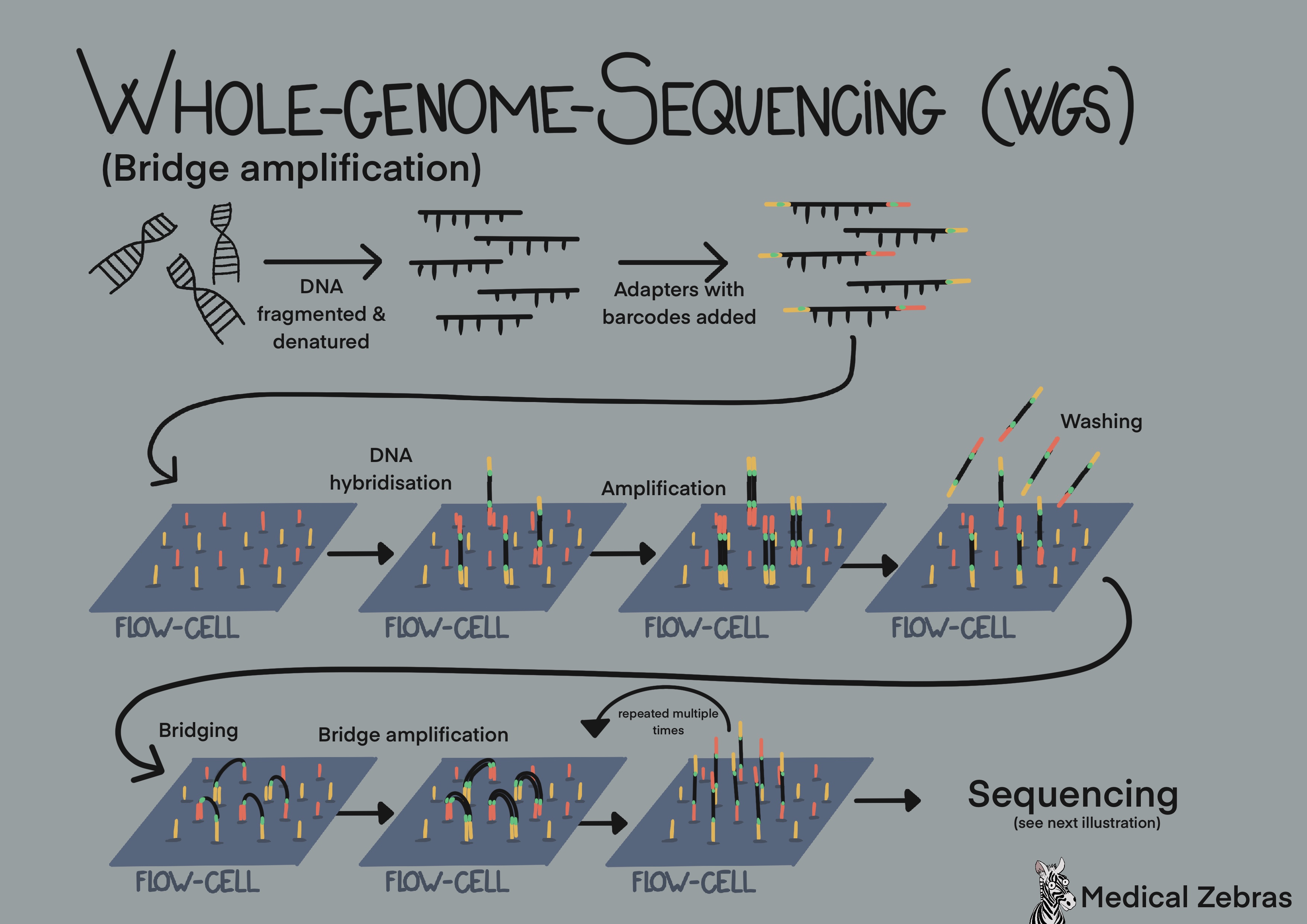
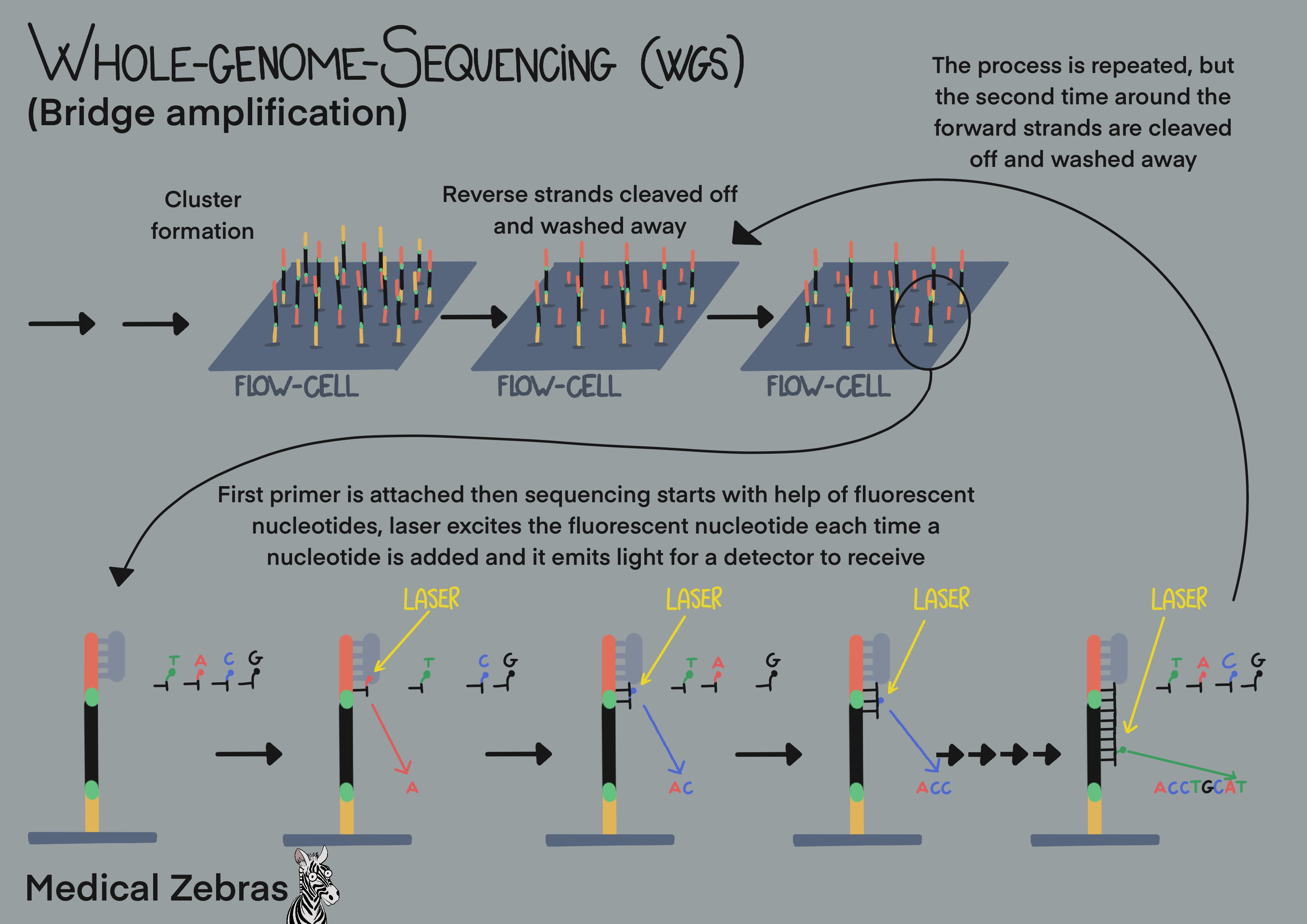
Bridge amplification (used by Illumina platforms): use so-called cluster generation and bridge amplification. First the DNA is fragmented in fragments of 200-300 bp, often done by sonication. Thereafter adaptars, short specific oligonucleotide sequences, are added to the free ends of the fragmented DNA, so the DNA can attach to the surface of the flow cell, a glass surface that has thousands of short oligonucleotide sequence complimentary to the adapters attached to it. Sometimes these adapters also contain specific barcodes. That way many samples can be pooled together (sequenced together in the same run) and identified later in the analyzing process. After the DNA fragments have attached (hybridized) to the flow cell, polymerase can attach to where the adapters are and elongate the DNA string. Denaturing separates the newly formed DNA from the old one, and the new fragment attaches to another adapter. Elongation ensues, thereafter denaturization. As there are adapters on both sides of the DNA fragments, the fragments can attach to the flow cell on both sides forming bridges. When the fragment is elongated in this shape, it is called bridge amplification and it is that way that most of the amplification gets done during amplification. When amplification is done, all available attachments on the flow cell are occupied by one strand. Now the reverse strands are removed, leaving only the forward strands occupying the flow cell. Fluorescently labelled nucleotides are added to the flow cells and a "photo" is snapped each time a nucleotide is added (laser excites fluorescent nucleotides that emit light). After the desired read length is attchieved, one amplification cycle is repeated and thereafter the forward strands are washed away, leaving only the reverse strands and sequencing is repeated with fluorecently labelled nucleotides. As all fragments have been sequenced, we end up with many DNA fragments that overlap to form a long sequence that is then compared to a reference genome. Different mapping programs/specific algorithms can be used to achieve this, the most known one is Bowtie2 (open-source) and Novoalign.
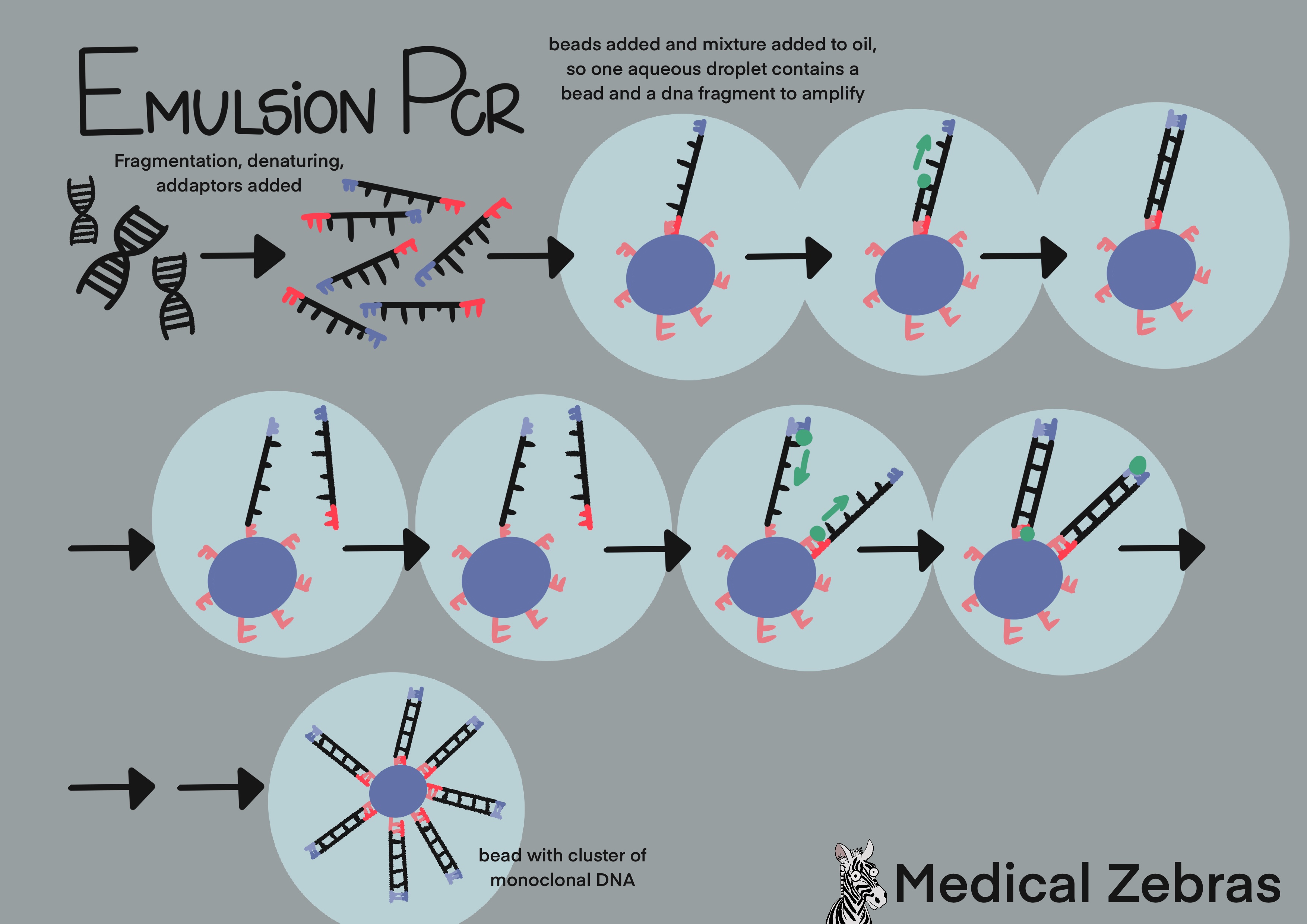
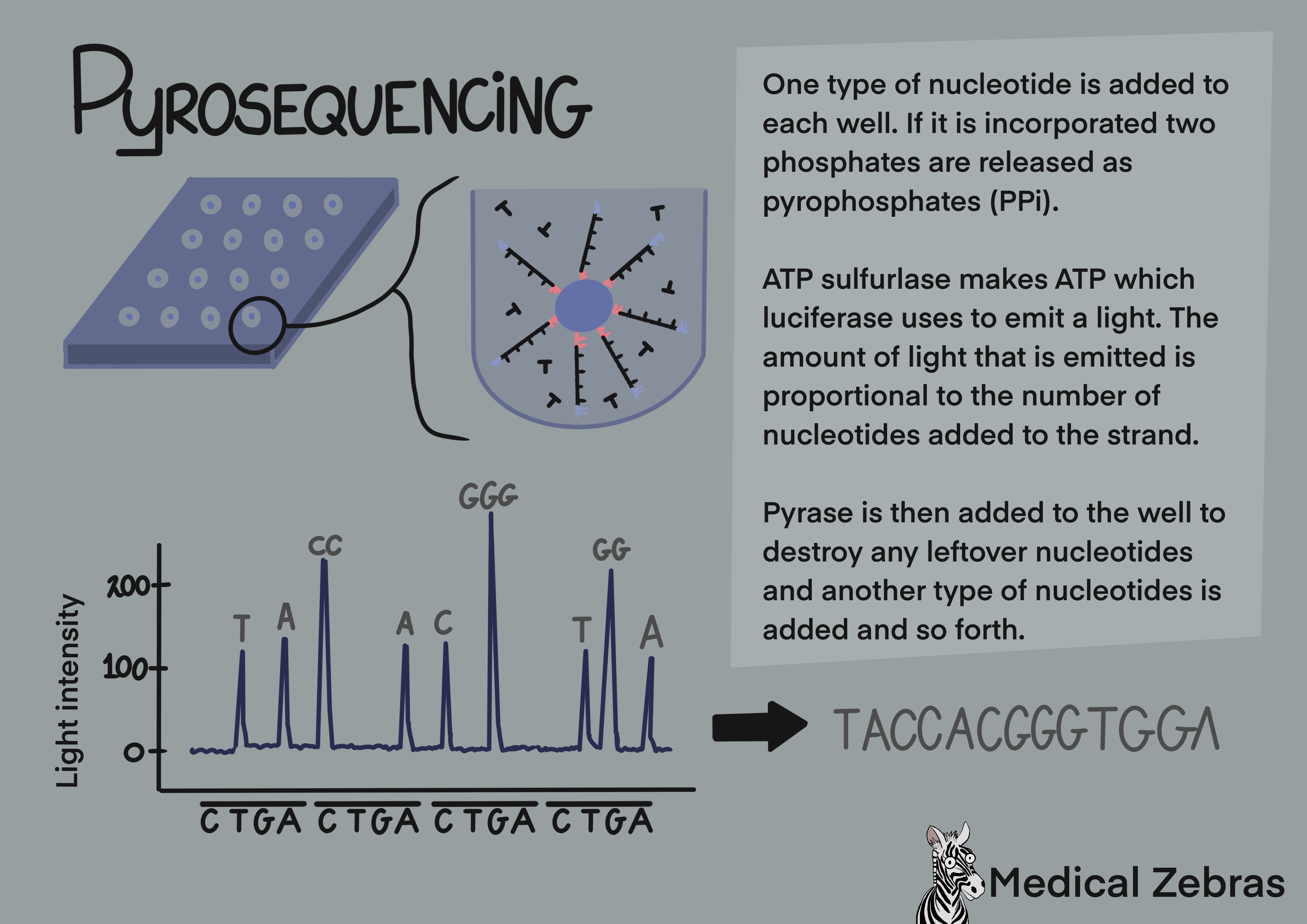
Emulsion PCR and Pyrosequencing (used by Ion Torrent): Here aqueous solution is used. This is a mix of beads with sequencing primers attached that the adapters can attach to, test DNA that has been fragmented and adapters attached to the free ends and reverse primers. The solution is mixed with oil where water droplets become entrapt in the oil containing individual DNA fragments and a bead. The DNA strand binds to the bead and through many cycles of amplification forms clusters of many copies of a single type of DNA fragment that is bound to it. The beads are then transferred to a sequencing plate containing many wells (one well for each bead), where sequencing takes place. Nucleotides are pumped into the wells, only one type at a time with washing and removal of nucleotides in between. If the right one is released into a well ATP is released as the nucleotide is incorporated into the newly formed DNA strand and luciferase enzyme uses it to produce a light signal for a computer to detect. This is called pyrosequencing. A downside of pyrosequencing is that it can be hard for the computers detector to distinguish a row of the same nucleotide, for example 7 T's from 8 T's, as they all attach in the same run and the difference in light strength measured can be hard to detect.
Bridge amplification (used by Illumina platforms): use so-called cluster generation and bridge amplification. First the DNA is fragmented in fragments of 200-300 bp, often done by sonication. Thereafter adaptars, short specific oligonucleotide sequences, are added to the free ends of the fragmented DNA, so the DNA can attach to the surface of the flow cell, a glass surface that has thousands of short oligonucleotide sequence complimentary to the adapters attached to it. Sometimes these adapters also contain specific barcodes. That way many samples can be pooled together (sequenced together in the same run) and identified later in the analyzing process. After the DNA fragments have attached (hybridized) to the flow cell, polymerase can attach to where the adapters are and elongate the DNA string. Denaturing separates the newly formed DNA from the old one, and the new fragment attaches to another adapter. Elongation ensues, thereafter denaturization. As there are adapters on both sides of the DNA fragments, the fragments can attach to the flow cell on both sides forming bridges. When the fragment is elongated in this shape, it is called bridge amplification and it is that way that most of the amplification gets done during amplification. When amplification is done, all available attachments on the flow cell are occupied by one strand. Now the reverse strands are removed, leaving only the forward strands occupying the flow cell. Fluorescently labelled nucleotides are added to the flow cells and a "photo" is snapped each time a nucleotide is added (laser excites fluorescent nucleotides that emit light). After the desired read length is attchieved, one amplification cycle is repeated and thereafter the forward strands are washed away, leaving only the reverse strands and sequencing is repeated with fluorecently labelled nucleotides. As all fragments have been sequenced, we end up with many DNA fragments that overlap to form a long sequence that is then compared to a reference genome. Different mapping programs/specific algorithms can be used to achieve this, the most known one is Bowtie2 (open-source) and Novoalign.
Emulsion PCR and Pyrosequencing (used by Ion Torrent): Here aqueous solution is used. This is a mix of beads with sequencing primers attached that the adapters can attach to, test DNA that has been fragmented and adapters attached to the free ends and reverse primers. The solution is mixed with oil where water droplets become entrapt in the oil containing individual DNA fragments and a bead. The DNA strand binds to the bead and through many cycles of amplification forms clusters of many copies of a single type of DNA fragment that is bound to it. The beads are then transferred to a sequencing plate containing many wells (one well for each bead), where sequencing takes place. Nucleotides are pumped into the wells, only one type at a time with washing and removal of nucleotides in between. If the right one is released into a well ATP is released as the nucleotide is incorporated into the newly formed DNA strand and luciferase enzyme uses it to produce a light signal for a computer to detect. This is called pyrosequencing. A downside of pyrosequencing is that it can be hard for the computers detector to distinguish a row of the same nucleotide, for example 7 T's from 8 T's, as they all attach in the same run and the difference in light strength measured can be hard to detect.
Pros: Genome-wide exon coverage. Results can be
obtained in one day. Cheaper than WGS.
Cons: Only covers known exons. Does not sequence repeats very well and are therefore not ideal for diagnosing repeat disorders (for example Fragile-X). Coverage can vary between runs. Coverage not as optimal as when only a couple of exons are sequenced. Challenging to identify large structural variants (inversions, translocations, insertion and deletion) as the DNA is sequenced in small fragments of 150-250bp. Not guarenteed that mitochondrial DNA is covered, depends on how the sample is prepped and what kits are used.
Examples of uses in the field
~ acutely sick infants/children and adults of unknown causes justifying a wide screen for potential genetic diseases.
~ clusters of symptoms that point to a genetic defect but where it is hard to rule out many genes.
Cons: Only covers known exons. Does not sequence repeats very well and are therefore not ideal for diagnosing repeat disorders (for example Fragile-X). Coverage can vary between runs. Coverage not as optimal as when only a couple of exons are sequenced. Challenging to identify large structural variants (inversions, translocations, insertion and deletion) as the DNA is sequenced in small fragments of 150-250bp. Not guarenteed that mitochondrial DNA is covered, depends on how the sample is prepped and what kits are used.
Examples of uses in the field
~ acutely sick infants/children and adults of unknown causes justifying a wide screen for potential genetic diseases.
~ clusters of symptoms that point to a genetic defect but where it is hard to rule out many genes.
Whole-genome-sequencing (WGS)
The test protocols, i.e. bridge amplification and emulsion PCR, are the
same as with WES described above. The difference with WGS is that all of
the DNA is added to the flow cell after fragmentation, not only the
exons.
Examples of uses in the field
~ Same as with WES or when looking specifically for intron or mitochondrial variants.
Pros: better coverage than WES, as it covers areas
around the exons as well as mitochondrial DNA and of course introns.
Cons: Same as with WES. Expensive. A significant chance of finding many variants of unknown significance (VUS), as intron functions are not well known today and most known genetic diseases are caused by patogenic mutations in exons. For this reason, with our current understanding, WGS maybe not adding alot of information over WES unless we are looking for mitochondrial or intron mutations. As with WES, it is challenging to identify large structural variants such as inversions, translocations, insertions and deletions since the DNA is sequenced in small fragments of 150-250bp.
Cons: Same as with WES. Expensive. A significant chance of finding many variants of unknown significance (VUS), as intron functions are not well known today and most known genetic diseases are caused by patogenic mutations in exons. For this reason, with our current understanding, WGS maybe not adding alot of information over WES unless we are looking for mitochondrial or intron mutations. As with WES, it is challenging to identify large structural variants such as inversions, translocations, insertions and deletions since the DNA is sequenced in small fragments of 150-250bp.
Examples of uses in the field
~ Same as with WES or when looking specifically for intron or mitochondrial variants.
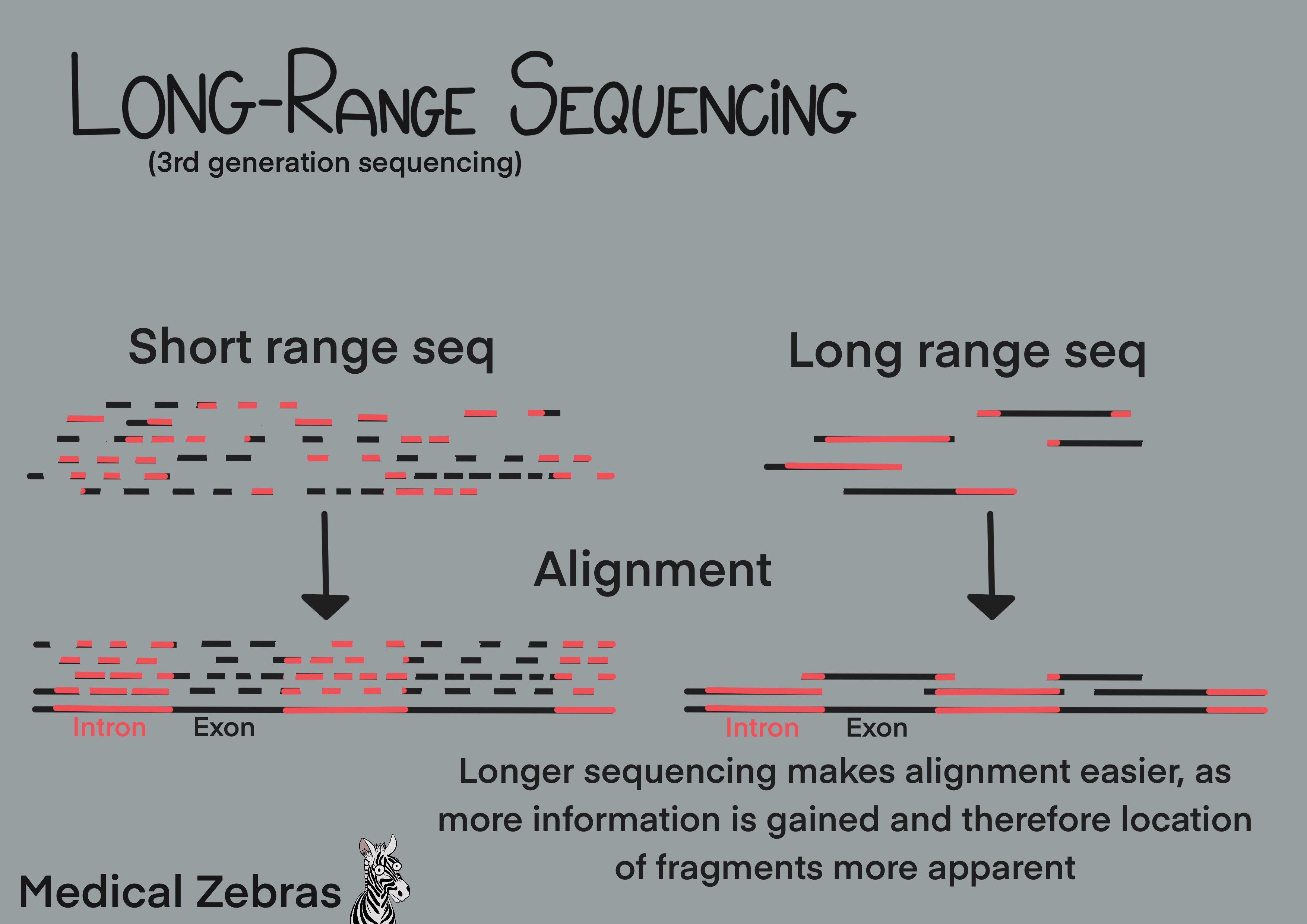
Long-range sequencing
Long-range sequencing allows for the sequencing of DNA fragments which
can be on average over 10kb. This extra length allows for better
resolution and easier aligning during analysis. This technique makes it
easier to sequence repetitive elements in genomes and allows for better
detection of large-scale changes in the genome. It even allows for
direct detection of DNA methylation (epigenetic modifications).
Pros: Better DNA coverage i.e. with less gaps, rows
of repeated basepairs are better sequenced (otherwise it is error
prone and hard to align), easier to sequence new unknown genomes. Can
detect large-scale changes in the genome (large inversions,
insertions, deletions). Better detection of pseudogenes as the
sequence around them is mapped simutaniously (and then you can
differentiate them from the functional gene that can have a nearly
identical sequence). Easier to determine parental origin of mutations.
Cons:Expensive. Higher error rates than short read(SR)-sequencing.
Examples of uses in the field (for example)
~ sequencing of new species or challenging genomes with highly repetitive sequences.
Cons:Expensive. Higher error rates than short read(SR)-sequencing.
Examples of uses in the field (for example)
~ sequencing of new species or challenging genomes with highly repetitive sequences.
Southern blot
Is a method for DNA gene detection analysis. DNA is cut into smaller
fragments using restriction endonucleases and thereafter are seperated
by electrophoresis on an agarose gel (similar to gel electrophoresis
after PCR). To be able to add probes for hybridization of the gene of
interest later, the DNA is denatured (DNA strands separated) by adding
an alkaline solution over the gel. After electrophoresis a nylon
membrane is placed onto the gel. The DNA in the gel is thereafter
transferred by capillary-action onto the membrane. The DNA sticks to the
membrane as it is positively charged and the DNA negatively charged. To
permanently attach the DNA to the membrane, it is exposed to UV light.
Probes that can attach to the specific sequence of interest are added to
the memebrane and left to hybridize to the DNA. To be able to detect the
bound probes they are lablled with a fluorescent or chromogenic dye. Now
the DNA fragments can be detected by X-ray(fluorescent probe) or colour
development (chromogenetic detection).
Southern blot can also be used to check for gene copy number. Lets say that a gene has couple of copy number variants (CNV). For each CNV, the restriction enzyme cuts differently as the area around the gene copies varies. Therefore we get different sizes of DNA fragments for each copy number variant which will appear on the membrane as separate bands when they are separated by length in the electrophoresis process.
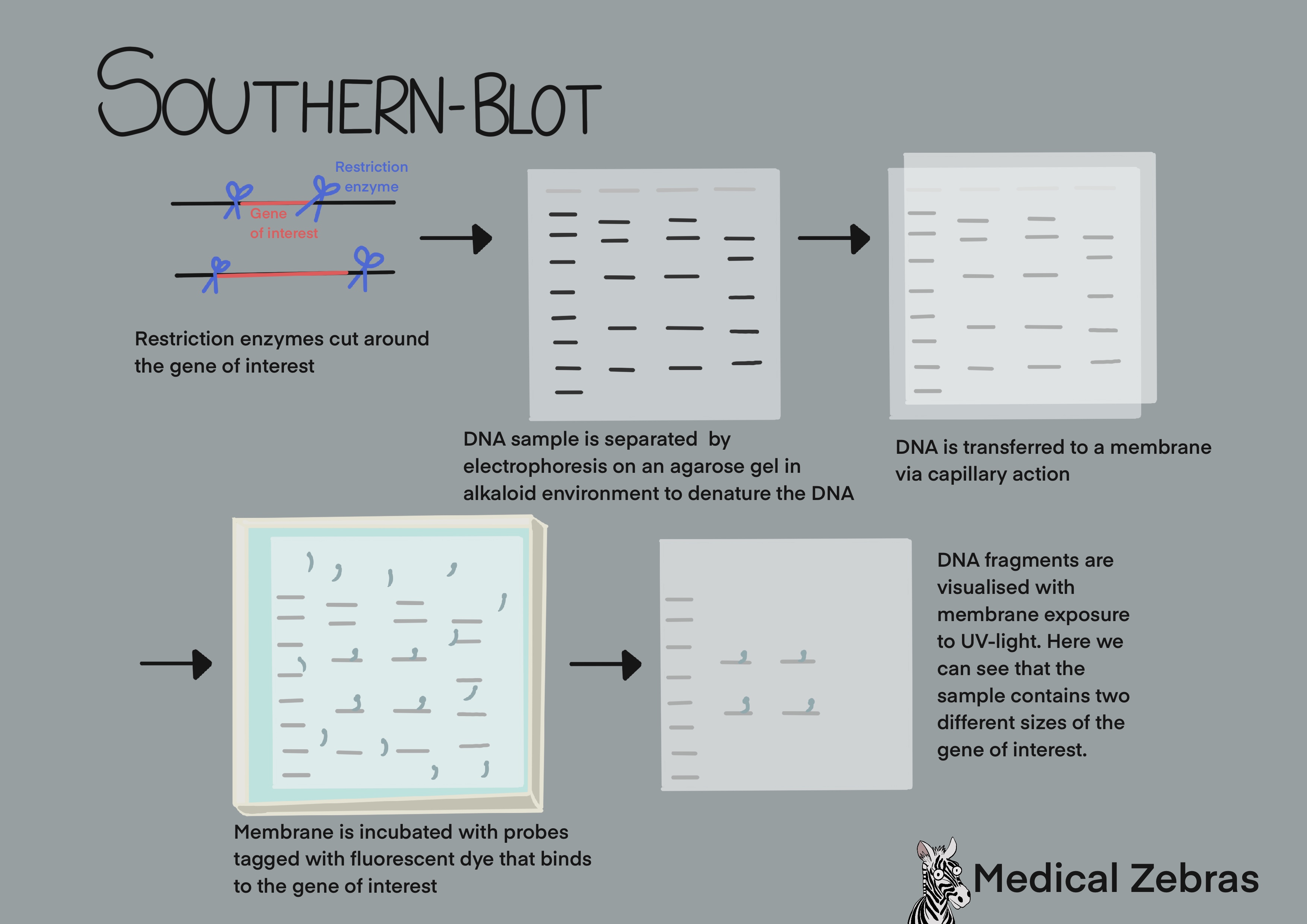
Southern blot can also be used to check for gene copy number. Lets say that a gene has couple of copy number variants (CNV). For each CNV, the restriction enzyme cuts differently as the area around the gene copies varies. Therefore we get different sizes of DNA fragments for each copy number variant which will appear on the membrane as separate bands when they are separated by length in the electrophoresis process.
Pros: Can be used to detect if gene is present or
not, and help with determining the copy number of a gene. Easy
methylation detection.
Cons: A time consuming and outdated method. Can only detect what the probes that are used can attach to.
Examples of uses in the field (for example)
~ Diagnosing Fragile X (over 200 CGG repeats in the FMR1 gene).
~ DNA fingerprinting in forensic science.
Cons: A time consuming and outdated method. Can only detect what the probes that are used can attach to.
Examples of uses in the field (for example)
~ Diagnosing Fragile X (over 200 CGG repeats in the FMR1 gene).
~ DNA fingerprinting in forensic science.
Triplet-Primed PCR
As the name implies, three primers are used for the PCR process. A
forward primer containing a fluorescent probe which binds to the region
before the repeat sequence starts. A second primer that binds within the
repeated region with a primer tail and a third primer able to attach to
the primer tail of the second primer. As the PCR reaction goes on, the
primer that binds to the repeat sequence, randomly attaches within the
repeat sequence resulting in a PCR product of variable length. The more
variation in the length of the product that is created, the larger the
repeat sequence is as the primer has more places to bind to. The output
of the PCR is visualised on an electropherogram which is generated with
capillary electrophoresis where the fragments are separated and
identified by size.
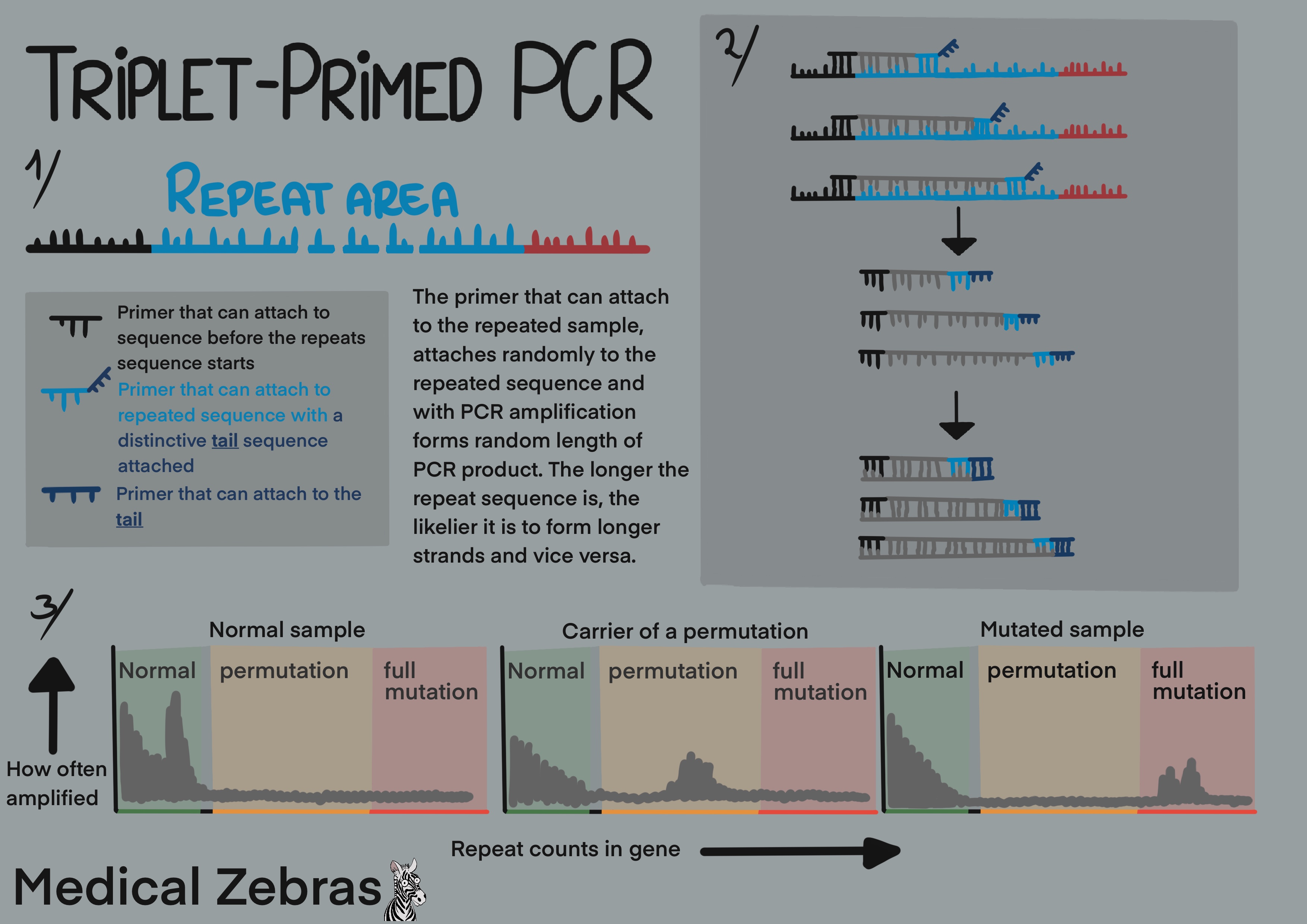
Pros: little DNA is required and it can even be used
in a single-cell setting, for example in preimplantation testing. Not
as labour intensive as Southern blotting.
Cons: Methylation testing cannot be done simutaniously (as would be optimal in the case of Fragile X diagnosis). Cannot accurately measure expansions, and therefore cannot give accurate answers, but instead provides a range.
Examples of uses in the field:
~ Diagnosing triple repeat disorders.
Cons: Methylation testing cannot be done simutaniously (as would be optimal in the case of Fragile X diagnosis). Cannot accurately measure expansions, and therefore cannot give accurate answers, but instead provides a range.
Examples of uses in the field:
~ Diagnosing triple repeat disorders.
Noninvasive prenatal testing (NIPT)
Noninvasive prenatal testing or NIPT is a genetic analysis used for the
diagnosis of chromosomal abnormalities, primarily trisomy 21, 18 and 13.
It is based on detecting so-called cell free fetal DNA (cffDNA). This
placental DNA and fetal DNA is free-floating DNA circulating in the
mothers blood stream which can be detected in the mothers blood as early
as from the 5th to 7th week of gestation. The amount of cffDNA increases
over time and from the 9th-10th week of gestation, enough of it is
present for its detection in a standard blood sample from the mother.
This DNA is isolated and is sequenced to a sufficient depth to identify
possible extra chromosomes. This will show up in the analysis as a
greater amount of the genes found on these chromosomes when compared to
the normal amount owing to there being more copies of them for the
polymerase to sequence.
After isolation of the free floating DNA from the blood, it can be analyzed in four different ways:
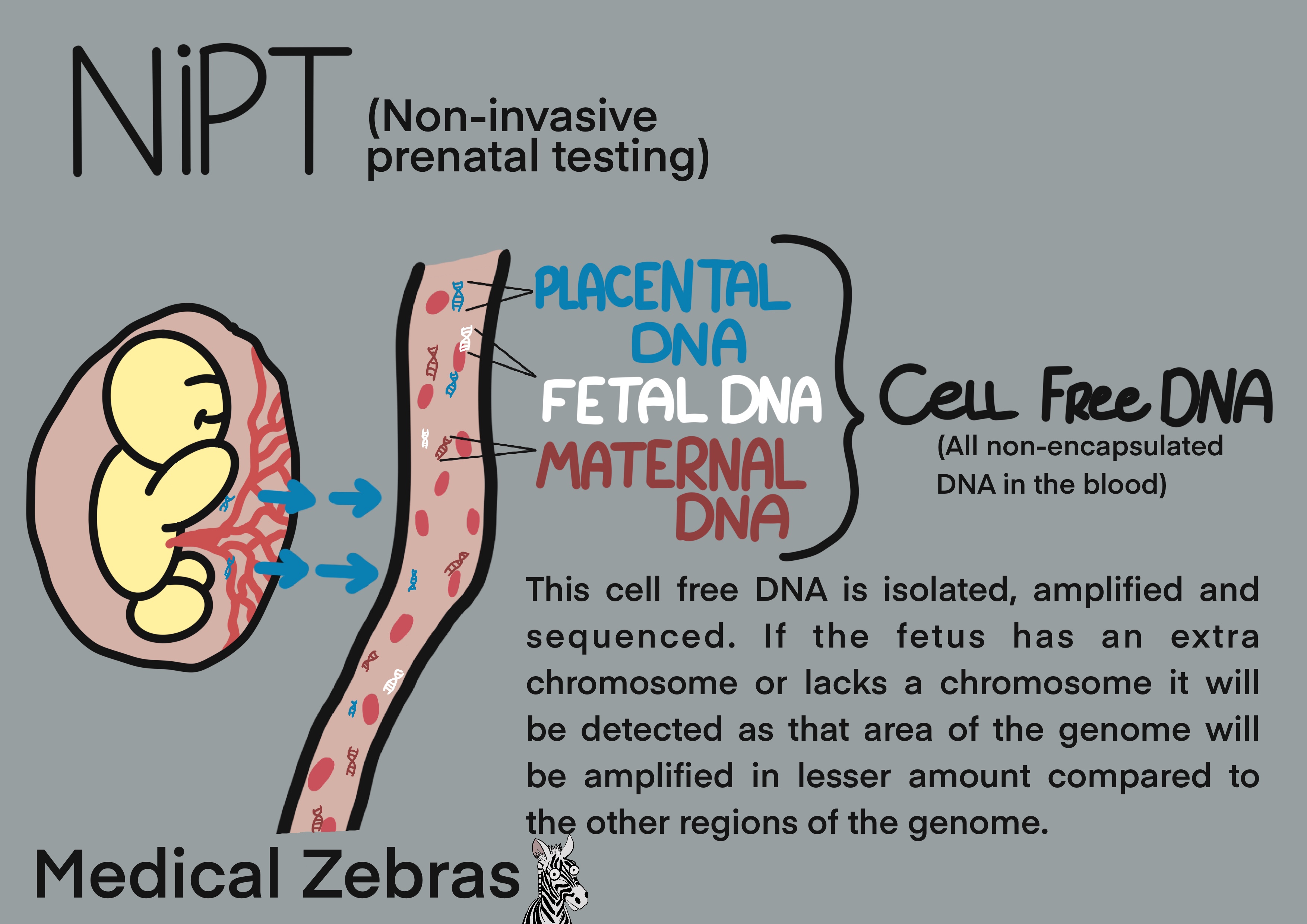
After isolation of the free floating DNA from the blood, it can be analyzed in four different ways:
- Whole-Genome-Sequencing
- SNP Analysis
- Microarray Analysis
- Rolling Circle Amplification
Pros: Less invasive testing compared to amniocentesis
and chorionic villus sampling.
Cons:
Multiple fetuses reduces the sensitivity and specificity of the test. Maternal or placental mosaicism can cause false positive results.
Uses in the field:
~ screen for aneuploidies
~ determine Rhesus typing to anticipate for possible problems with hemolytic desease of the newborn
Cons:
Multiple fetuses reduces the sensitivity and specificity of the test. Maternal or placental mosaicism can cause false positive results.
Uses in the field:
~ screen for aneuploidies
~ determine Rhesus typing to anticipate for possible problems with hemolytic desease of the newborn
Multiplex ligation-dependent probe amplification assay (MLPA)
Multiplex ligation-dependet probe amplification assay or MLPA is used to
detect genetic deletions and duplications. It is a form of multiplex
PCR, where many primer pairs are used simutaniously in a sample.
However, in MLPA, the primers pairs, or probe pairs, are designed to
hybridize next to one another on the same strand of DNA. This means that
if they are able to attach to the DNA, a ligase catalizes a covalent
bond between the probe pair forming a strand of a defined length. As the
ligase is specific to the region of interest, it is unable to ligate the
probes if there is a mismatch present at the ligation site. Because of
this specificity, MLPA can be used to identify a gene from a pseudogene,
i.e. a nonfunctioning DNA segment which resembles another functional
gene, or to detect a point mutatio in a gene. Thereafter the sample is
amplified using fluorecently labelled forward primers. Using capillary
electrophoresis the DNA strands are separated and identified from their
specified length. Comparing fluorescent intensity from the test sample
and control, a ratio is calculated and the number of gene copies
determined. If both copies of the diploid gene are present, a ratio of
1:1 between the sample and control is expected. If the sample contains
an extra copy of a gene, a ratio of 1:1.5 or greater would be expected.
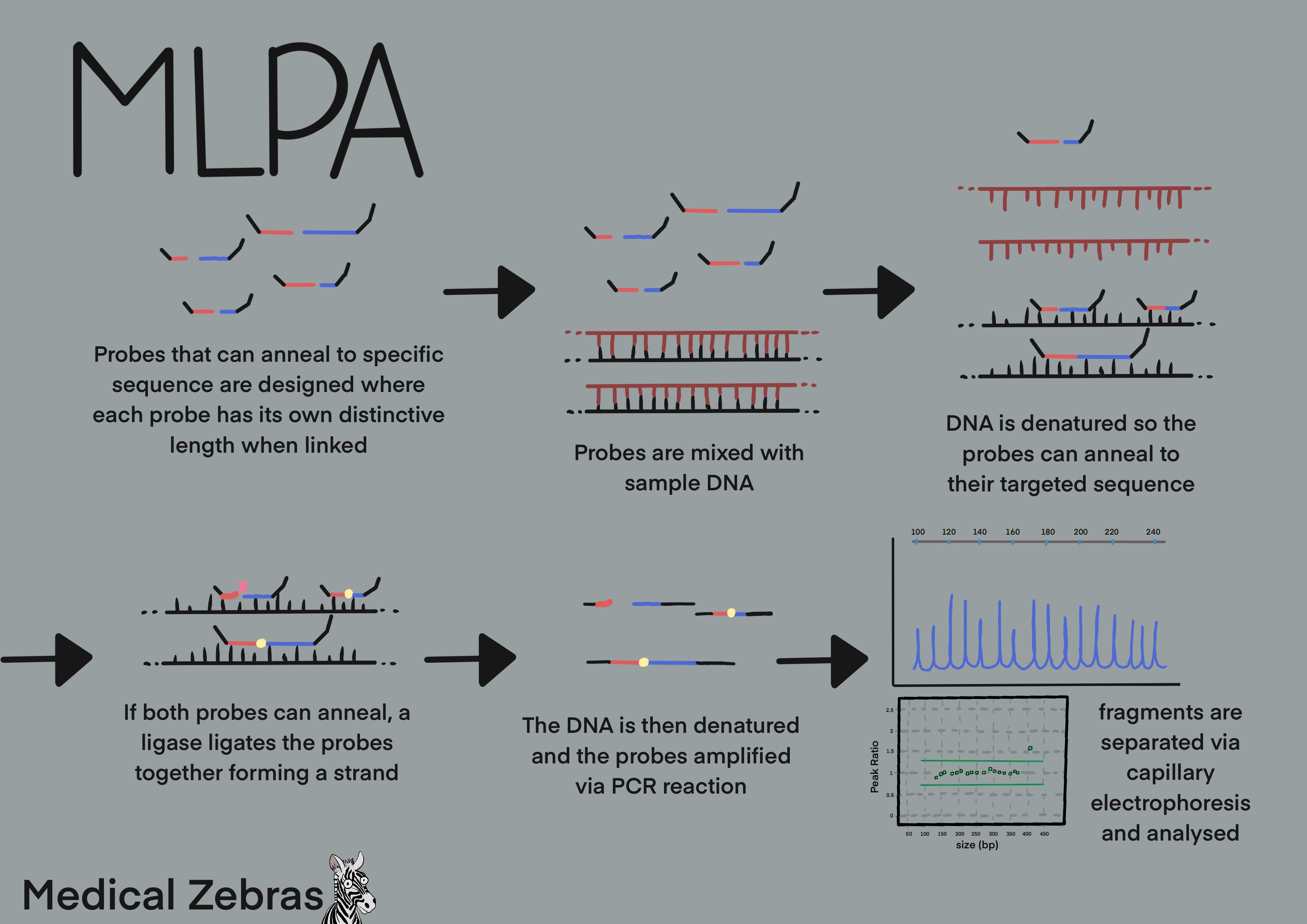
Pros: good analysis to Detect pseudogenes. For
example,determining CNVs in the PMS2 gene. The gene PMS2 has a
pseudogene, making the detection of CNVs in the gene via WES/WGS
analysis difficult. This can be overcome with MLPA.
Cons: Any mismatch in the probes target site can theoratically affect the probes binding and therefore affect its signal in analysis. Mismatches can therefore disguise as deletions. Probes are often designed to attach to areas where known SNPs and polymorphisms are not present.
Cons: Any mismatch in the probes target site can theoratically affect the probes binding and therefore affect its signal in analysis. Mismatches can therefore disguise as deletions. Probes are often designed to attach to areas where known SNPs and polymorphisms are not present.